Synthesis, Characterization and Development of Rapid, Sensitive Methods for the Detection of Potent Synthetic Opiates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Review of Synthetic Fentanyl Metabolism and the Metabolism of Select Synthetic Fentanyl Analogues
A review of synthetic fentanyl metabolism and the metabolism of select synthetic fentanyl analogues Gerard Lee A thesis submitted for the completion of a degree in Master of Forensic Science (Professional Practice) in The School of Veterinary and Life Sciences Murdoch University Supervisors: Associate Professor James Speers (Murdoch) Associate Professor Bob Mead (Murdoch) Semester 1, 2019 i Declaration I declare that this thesis does not contain any material submitted previously for the award of any other degree or diploma at any university or other tertiary institution. Furthermore, to the best of my knowledge, it does not contain any material previously published or written by another individual, except where due reference has been made in the text. Finally, I declare that all reported experimentations performed in this research were carried out by myself, except that any contribution by others, with whom I have worked is explicitly acknowledged. ii Acknowledgements I would like to thank my supervisor Bob Mead for his time guiding me and providing feedback on this endeavour. He has been a great help providing insights and advice from when I started university at Murdoch until now for which I am extremely grateful. To James Speers, thank you for helping me find a direction for this project when I started out. And finally, to my family and friends for their encouragement and support. iii Table of Contents Title page. ...............................................................................................................................I -

18 December 2020 – to Date)
(18 December 2020 – to date) MEDICINES AND RELATED SUBSTANCES ACT 101 OF 1965 (Gazette No. 1171, Notice No. 1002 dated 7 July 1965. Commencement date: 1 April 1966 [Proc. No. 94, Gazette No. 1413] SCHEDULES Government Notice 935 in Government Gazette 31387 dated 5 September 2008. Commencement date: 5 September 2008. As amended by: Government Notice R1230 in Government Gazette 32838 dated 31 December 2009. Commencement date: 31 December 2009. Government Notice R227 in Government Gazette 35149 dated 15 March 2012. Commencement date: 15 March 2012. Government Notice R674 in Government Gazette 36827 dated 13 September 2013. Commencement date: 13 September 2013. Government Notice R690 in Government Gazette 36850 dated 20 September 2013. Commencement date: 20 September 2013. Government Notice R104 in Government Gazette 37318 dated 11 February 2014. Commencement date: 11 February 2014. Government Notice R352 in Government Gazette 37622 dated 8 May 2014. Commencement date: 8 May 2014. Government Notice R234 in Government Gazette 38586 dated 20 March 2015. Commencement date: 20 March 2015. Government Notice 254 in Government Gazette 39815 dated 15 March 2016. Commencement date: 15 March 2016. Government Notice 620 in Government Gazette 40041 dated 3 June 2016. Commencement date: 3 June 2016. Prepared by: Page 2 of 199 Government Notice 748 in Government Gazette 41009 dated 28 July 2017. Commencement date: 28 July 2017. Government Notice 1261 in Government Gazette 41256 dated 17 November 2017. Commencement date: 17 November 2017. Government Notice R1098 in Government Gazette 41971 dated 12 October 2018. Commencement date: 12 October 2018. Government Notice R1262 in Government Gazette 42052 dated 23 November 2018. -

(12) United States Patent (10) Patent No.: US 9,687,445 B2 Li (45) Date of Patent: Jun
USOO9687445B2 (12) United States Patent (10) Patent No.: US 9,687,445 B2 Li (45) Date of Patent: Jun. 27, 2017 (54) ORAL FILM CONTAINING OPIATE (56) References Cited ENTERC-RELEASE BEADS U.S. PATENT DOCUMENTS (75) Inventor: Michael Hsin Chwen Li, Warren, NJ 7,871,645 B2 1/2011 Hall et al. (US) 2010/0285.130 A1* 11/2010 Sanghvi ........................ 424/484 2011 0033541 A1 2/2011 Myers et al. 2011/0195989 A1* 8, 2011 Rudnic et al. ................ 514,282 (73) Assignee: LTS Lohmann Therapie-Systeme AG, Andernach (DE) FOREIGN PATENT DOCUMENTS CN 101703,777 A 2, 2001 (*) Notice: Subject to any disclaimer, the term of this DE 10 2006 O27 796 A1 12/2007 patent is extended or adjusted under 35 WO WOOO,32255 A1 6, 2000 U.S.C. 154(b) by 338 days. WO WO O1/378O8 A1 5, 2001 WO WO 2007 144080 A2 12/2007 (21) Appl. No.: 13/445,716 (Continued) OTHER PUBLICATIONS (22) Filed: Apr. 12, 2012 Pharmaceutics, edited by Cui Fude, the fifth edition, People's Medical Publishing House, Feb. 29, 2004, pp. 156-157. (65) Prior Publication Data Primary Examiner — Bethany Barham US 2013/0273.162 A1 Oct. 17, 2013 Assistant Examiner — Barbara Frazier (74) Attorney, Agent, or Firm — ProPat, L.L.C. (51) Int. Cl. (57) ABSTRACT A6 IK 9/00 (2006.01) A control release and abuse-resistant opiate drug delivery A6 IK 47/38 (2006.01) oral wafer or edible oral film dosage to treat pain and A6 IK 47/32 (2006.01) substance abuse is provided. -

(12) United States Patent (10) Patent No.: US 8,231,900 B2 Palmer Et Al
USOO823 1900B2 (12) United States Patent (10) Patent No.: US 8,231,900 B2 Palmer et al. (45) Date of Patent: *Jul. 31, 2012 (54) SMALL-VOLUME ORAL TRANSMUCOSAL 4,873,076 A 10, 1989 Fishman et al. DOSAGE 4,880,634 A 1 1/1989 Speiser et al. 5,080,903. A 1/1992 Ayache 5,112,616 A 5/1992 McCartv et al. (75) Inventors: Pamela Palmer, San Francisco, CA 5, 122,127 A 6, 1992 St. (US); Thomas Schreck, Portola Valley, 5,132,114 A 7/1992 Stanley CA (US); Stelios Tzannis, Newark, CA 5,178,878 A 1/1993 Wehling (US); Larry Hamel, Mountain View, CA 5,223,264 A 6, 1993 Wehling et al. O O 5,236,714 A 8, 1993 Lee (US); Andrew I. Poutiatine, San 5,288.497 A 2, 1994 Stanley Anselmo, CA (US) 5,288.498 A 2/1994 Stanley 5,296,234 A 3/1994 Hadaway (73) Assignee: Acelrx Pharmaceutical, Inc., Redwood 5,348,158 A 9, 1994 Honan et al. City, CA (US) 5,489,689 A * 2/1996 Mathew ........................ 546,242 s 5,507,277 A 4, 1996 Rubsamen et al. (*) Notice: Subject to any disclaimer, the term of this 3.68 A SE Exitl patent is extended or adjusted under 35 5,710,551 A 1/1998 Ridgeway et al. U.S.C. 154(b) by 229 days. 5,724,957 A 3, 1998 RubSamen et al. 5,735,263 A 4/1998 RubSamen et al. This patent is Subject to a terminal dis- 5,752,620 A 5/1998 Pearson claimer. -

Recommended Methods for the Identification and Analysis of Fentanyl and Its Analogues in Biological Specimens
Recommended methods for the Identification and Analysis of Fentanyl and its Analogues in Biological Specimens MANUAL FOR USE BY NATIONAL DRUG ANALYSIS LABORATORIES Laboratory and Scientific Section UNITED NATIONS OFFICE ON DRUGS AND CRIME Vienna Recommended Methods for the Identification and Analysis of Fentanyl and its Analogues in Biological Specimens MANUAL FOR USE BY NATIONAL DRUG ANALYSIS LABORATORIES UNITED NATIONS Vienna, 2017 Note Operating and experimental conditions are reproduced from the original reference materials, including unpublished methods, validated and used in selected national laboratories as per the list of references. A number of alternative conditions and substitution of named commercial products may provide comparable results in many cases. However, any modification has to be validated before it is integrated into laboratory routines. ST/NAR/53 Original language: English © United Nations, November 2017. All rights reserved. The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. Mention of names of firms and commercial products does not imply the endorse- ment of the United Nations. This publication has not been formally edited. Publishing production: English, Publishing and Library Section, United Nations Office at Vienna. Acknowledgements The Laboratory and Scientific Section of the UNODC (LSS, headed by Dr. Justice Tettey) wishes to express its appreciation and thanks to Dr. Barry Logan, Center for Forensic Science Research and Education, at the Fredric Rieders Family Founda- tion and NMS Labs, United States; Amanda L.A. -

Understanding and Challenging the Drugs: Chemistry and Toxicology
UNDERSTANDING AND CHALLENGING THE DRUGS: CHEMISTRY AND TOXICOLOGY Presenter: • Dr. Jasmine Drake, Graduate Program Director and Assistant Professor, Administration of Justice Department, Barbara Jordan-Mickey Leland School of Public Affairs, Texas Southern University NACDL Training Defending Drug Overdose Homicides in Pennsylvania Penn State Harrisburg, Middletown, PA November 6th, 2019 11:30- 12:45 p.m. Understanding & Challenging the Drugs: Chemistry & Toxicology Dr. Jasmine Drake, Forensic Science Learning Laboratory, Texas Southern University I. Opioid Drug Classifications A. Types of Opioids B. Classic vs. Synthetic C. Toxicology of Opioids 1) How opioids interact with the body 2) Addiction (psychological vs. physiological II. New Classes of Drugs A. Emerging Threats B. Potency III. National Trends in Opioid Overdose Deaths in the U.S. A. Based on State B. Ethnicity C. Drug-Type (prescription vs. fentanyl vs. heroin) IV. Trends of Opioid Overdose Deaths in Philadelphia A. Based on Ethnicity B. Drug Type (prescription vs. fentanyl vs. heroin) V. Legal Considerations to the Opioid Epidemic A. Punitive Measures vs. Rehabilitative Treatment B. Progressive Jurisdictions Nationwide C. New Legal Measures in Philadelphia VI. Toxicology Reports A. What’s in the report? B. Key Aspects of the Tox Report C. Terminology D. Evaluating and Interpreting the data? E. Questions and considerations. VII. Conclusion and Discussion A. Case Specific Examples B. Sample Toxicology Reports The Opioid Epidemic: What labs have to do with it? Ewa King, Ph.D. Associate Director of Health RIDOH State Health Laboratories Analysis. Answers. Action. www.aphl.org Overview • Overdose trends • Opioids and their effects • Analytical testing approaches • Toxicology laboratories Analysis. Answers. Action. -
Comprehensive Multi-Analytical Screening Of
COMPREHENSIVE MULTI-ANALYTICAL SCREENING OF DRUGS OF ABUSE, INCLUDING NEW PSYCHOACTIVE SUBSTANCES, IN URINE WITH BIOCHIP ARRAYS APPLIED TO THE EVIDENCE ANALYSER Darragh J., Keery L., Keenan R., Stevenson C., Norney G., Benchikh M.E., Rodríguez M.L., McConnell R. I., FitzGerald S.P. Randox Toxicology Ltd., Crumlin, United Kingdom e-mail: [email protected] Introduction Biochip array technology allows the simultaneous detection of multiple drugs from a single undivided sample, which This study summarises the analytical performance of three different biochip arrays applied to the screening of increases the screening capacity and the result output per sample. Polydrug consumption can be detected and by acetylfentanyl, AH-7921, amphetamine, barbiturates, benzodiazepines (including etizolam and clonazepam), incorporating new immunoassays on the biochip surface, this technology has the capacity to adapt to the new trends benzoylecgonine/cocaine, benzylpiperazines, buprenorphine, cannabinoids, carfentanil, dextromethorphan, fentanyl, in the drug market. furanylfentanyl, meprobamate, mescaline, methamphetamine, methadone, mitragynine, MT-45, naloxone, ocfentanyl, opioids, opiates, oxycodone, phencyclidine, phenylpiperazines, salvinorin, sufentanil, synthetic cannabinoids (JWH-018, UR-144, AB-PINACA, AB-CHMINACA), synthetic cathinones [mephedrone, methcathinone, alpha- pyrrolidinopentiophenone (alpha-PVP)], tramadol, tricyclic antidepressants, U-47700, W-19, zolpidem. Methodology Three different biochip arrays were used (DOA ULTRA, -

Critical Review Report: CROTONYLFENTANYL
Critical Review Report: CROTONYLFENTANYL Expert Committee on Drug Dependence Forty-second Meeting Geneva, 21-25 October 2019 This report contains the views of an international group of experts, and does not necessarily represent the decisions or the stated policy of the World Health Organization 42nd ECDD (2019): Crotonylfentanyl © World Health Organization 2019 All rights reserved. nd This is an advance copy distributed to the participants of the 42 Expert Committee on Drug Dependence, before it has been formally published by the World Health Organization. The document may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in any form or by any means without the permission of the World Health Organization. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted and dashed lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. The World Health Organization does not warrant that the information contained in this publication is complete and correct and shall not be liable for any damages incurred as a result of its use. -

The Synthetic Opioid N-Phenyl-N
Identification of a new psychoactive substance in seized material The synthetic opioid N-phenyl-N-[1-(2-phenethyl)piperidin-4-yl]prop-2-enamide (Acrylfentanyl) Breindahl, Torben; Kimergård, Andreas; Andreasen, Mette Findal; Pedersen, Daniel Sejer Published in: Drug Testing and Analysis DOI: 10.1002/dta.2046 Publication date: 2017 Document version Publisher's PDF, also known as Version of record Citation for published version (APA): Breindahl, T., Kimergård, A., Andreasen, M. F., & Pedersen, D. S. (2017). Identification of a new psychoactive substance in seized material: The synthetic opioid N-phenyl-N-[1-(2-phenethyl)piperidin-4-yl]prop-2-enamide (Acrylfentanyl). Drug Testing and Analysis, 9(3), 415-422. https://doi.org/10.1002/dta.2046 Download date: 28. Sep. 2021 Drug Testing Research article and Analysis Received: 28 June 2016 Revised: 21 July 2016 Accepted: 26 July 2016 Published online in Wiley Online Library: 24 August 2016 (www.drugtestinganalysis.com) DOI 10.1002/dta.2046 Identification of a new psychoactive substance in seized material: the synthetic opioid N- phenyl-N-[1-(2-phenethyl)piperidin-4-yl]prop- 2-enamide (Acrylfentanyl) Torben Breindahl,a* Andreas Kimergård,b Mette Findal Andreasenc and Daniel Sejer Pedersend Among the new psychoactive substances (NPS) that have recently emerged on the market, many of the new synthetic opioids have shown to be particularly harmful. A new synthetic analogue of fentanyl, N-phenyl-N-[1-(2-phenethyl)piperidin-4-yl]prop- 2-enamide (acrylfentanyl), was identified in powder from a seized capsule found at a forensic psychiatric ward in Denmark. Gas chromatography with mass spectrometry (GC-MS) identified a precursor to synthetic fentanyls, N-phenyl-1-(2-phenylethyl) piperidin-4-amine; however, the precursor 1-(2-phenethyl)piperidin-4-one, was not detected. -

Model Scheduling New/Novel Psychoactive Substances Act (Third Edition)
Model Scheduling New/Novel Psychoactive Substances Act (Third Edition) July 1, 2019. This project was supported by Grant No. G1799ONDCP03A, awarded by the Office of National Drug Control Policy. Points of view or opinions in this document are those of the author and do not necessarily represent the official position or policies of the Office of National Drug Control Policy or the United States Government. © 2019 NATIONAL ALLIANCE FOR MODEL STATE DRUG LAWS. This document may be reproduced for non-commercial purposes with full attribution to the National Alliance for Model State Drug Laws. Please contact NAMSDL at [email protected] or (703) 229-4954 with any questions about the Model Language. This document is intended for educational purposes only and does not constitute legal advice or opinion. Headquarters Office: NATIONAL ALLIANCE FOR MODEL STATE DRUG 1 LAWS, 1335 North Front Street, First Floor, Harrisburg, PA, 17102-2629. Model Scheduling New/Novel Psychoactive Substances Act (Third Edition)1 Table of Contents 3 Policy Statement and Background 5 Highlights 6 Section I – Short Title 6 Section II – Purpose 6 Section III – Synthetic Cannabinoids 13 Section IV – Substituted Cathinones 19 Section V – Substituted Phenethylamines 23 Section VI – N-benzyl Phenethylamine Compounds 25 Section VII – Substituted Tryptamines 28 Section VIII – Substituted Phenylcyclohexylamines 30 Section IX – Fentanyl Derivatives 39 Section X – Unclassified NPS 43 Appendix 1 Second edition published in September 2018; first edition published in 2014. Content in red bold first added in third edition. © 2019 NATIONAL ALLIANCE FOR MODEL STATE DRUG LAWS. This document may be reproduced for non-commercial purposes with full attribution to the National Alliance for Model State Drug Laws. -

TESTIMONY of CASEY OWEN DURST Director of Field Operations
TESTIMONY OF CASEY OWEN DURST Director of Field Operations Baltimore Field Office Office of Field Operations U.S. Customs and Border Protection Department of Homeland Security For a Hearing BEFORE U.S. House of Representatives Committee on Homeland Security Subcommittee on Oversight and Management Efficiency ON “Combating Opioid Smuggling” June 19, 2018 Harrisburg, Pennsylvania Introduction Chairman Perry, Ranking Member Correa, and distinguished Members of the Subcommittee, thank you for the opportunity to appear today to discuss the role of U.S. Customs and Border Protection (CBP) in combating the flow of opioids, including synthetic opioids such as fentanyl, into the United States. The opioid crisis is one of the most important, complex, and difficult challenges our Nation faces today, and was declared a National Emergency by President Donald Trump in October of last year.1 As America’s unified border agency, CBP plays a critical role in preventing illicit narcotics, including opioids, from reaching the American public. CBP leverages targeting and intelligence- driven strategies, and works in close coordination with our partners as part of our multi-layered, risk-based approach to enhance the security of our borders and our country. This layered approach reduces our reliance on any single point or program, and extends our zone of security outward, ensuring our physical border is not the first or last line of defense, but one of many. Opioid Trends, Interdictions, and Challenges In Fiscal Year (FY) 2018 to-date, the efforts of Office of Field Operations (OFO) and U.S. Border Patrol (USBP) personnel resulted in the seizure of more than 545,000 lbs. -
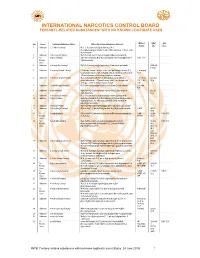
INTERNATIONAL NARCOTICS CONTROL BOARD FENTANYL-RELATED Substancesa with NO KNOWN LEGITIMATE USES
INTERNATIONAL NARCOTICS CONTROL BOARD a FENTANYL-RELATED SUBSTANCES WITH NO KNOWN LEGITIMATE USES Abbrev- CAS Intl No. Uses b Common Substance Name Other/ Alternative Substance Name(s) c iations No.d Ctrl.e 1 Unknown 2,2'-difluorofentanyl N-(1-(2-fluorophenethyl)piperidin-4-yl)-N-(2- fluorophenyl)propionamide; 2'-ortho-difluorofentanyl; 2'-fluoro ortho- fluorofentanyl 2 Unknown 2-fluoro butyrfentanyl N-(2-fluorophenyl)-N-(1-phenethylpiperidin-4-yl) butyramide 3 No 2-fluorofentanyl ortho-fluorofentanyl; N-(2-fluorophenyl)-N-(1-phenethylpiperidin-4- 2-FF; o-FF Known yl)propionamide Uses 4 Unknown 2-furanylethyl fentanyl N-[1-[2-(2-furanyl)ethyl]-4-piperidinyl]-N-phenyl-propanamide 1443-49- 8 (HCl) 5 Unknown 2-isopropylfuranyl fentanyl 2-isopropyl furanyl fentanyl; ortho-isopropyl furanyl fentanyl; N-(2- 2-isopropyl isopropylphenyl)-N-(1-phenethylpiperidin-4-yl)furan-2-carboxamide; Fu-F 2-Furanylfentanyl ortho-2-isopropylphenyl analogue 6 Unknown 2-methoxy furanyl fentanyl N-(2-methoxyphenyl)-N-[1-(2-phenylethyl)-4-piperidinyl]-2- 2-methoxy 101343- furancarboxamide; 2-Furanylfentanyl ortho-2-methoxyphenyl FuF; 2-Meo- 50-4 analogue; ortho-methoxy furanyl fentanyl FuF 7 Unknown 2-methyl furanyl fentanyl N-(1-phenethylpiperidin-4-yl)-N-(o-tolyl)furan-2-carboxamide 2-methyl FuF 8 Unknown 3-allyl fentanyl N-phenyl-N-[1-(2-phenylethyl)-3-(prop-2-en-1-yl)piperidin-4- 82208- yl]propanamide 84-2 9 Unknown 3-fluoro butyrfentanyl N-(3-fluorophenyl)-N-(1-phenethylpiperidin-4-yl) butyramide 10 Unknown 3-fluorofentanyl meta-fluorofentanyl; N-(3-fluorophenyl)-N-(1-phenethylpiperidin-4-