Donohue Syndrome EN
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstracts from the 9Th Biennial Scientific Meeting of The
International Journal of Pediatric Endocrinology 2017, 2017(Suppl 1):15 DOI 10.1186/s13633-017-0054-x MEETING ABSTRACTS Open Access Abstracts from the 9th Biennial Scientific Meeting of the Asia Pacific Paediatric Endocrine Society (APPES) and the 50th Annual Meeting of the Japanese Society for Pediatric Endocrinology (JSPE) Tokyo, Japan. 17-20 November 2016 Published: 28 Dec 2017 PS1 Heritable forms of primary bone fragility in children typically lead to Fat fate and disease - from science to global policy a clinical diagnosis of either osteogenesis imperfecta (OI) or juvenile Peter Gluckman osteoporosis (JO). OI is usually caused by dominant mutations affect- Office of Chief Science Advsor to the Prime Minister ing one of the two genes that code for two collagen type I, but a re- International Journal of Pediatric Endocrinology 2017, 2017(Suppl 1):PS1 cessive form of OI is present in 5-10% of individuals with a clinical diagnosis of OI. Most of the involved genes code for proteins that Attempts to deal with the obesity epidemic based solely on adult be- play a role in the processing of collagen type I protein (BMP1, havioural change have been rather disappointing. Indeed the evidence CREB3L1, CRTAP, LEPRE1, P4HB, PPIB, FKBP10, PLOD2, SERPINF1, that biological, developmental and contextual factors are operating SERPINH1, SEC24D, SPARC, from the earliest stages in development and indeed across generations TMEM38B), or interfere with osteoblast function (SP7, WNT1). Specific is compelling. The marked individual differences in the sensitivity to the phenotypes are caused by mutations in SERPINF1 (recessive OI type obesogenic environment need to be understood at both the individual VI), P4HB (Cole-Carpenter syndrome) and SEC24D (‘Cole-Carpenter and population level. -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

“Leprechaunism” with a Novel Mutation in the Insulin Receptor Gene
Case Report A case of Donohue syndrome “Leprechaunism” with a novel mutation in the insulin receptor gene Birgül Kirel1, Özkan Bozdağ2, Pelin Köşger2, Sultan Durmuş Aydoğdu2, Eylem Alıncak2, Neslihan Tekin3 1Osmangazi University, Faculty of Medicine, Department of Pediatrics, Division of Pediatric Endocrinology, Eskişehir, Turkey 2Osmangazi University, Faculty of Medicine, Department of Pediatrics, Division of General Pediatrics, Eskişehir, Turkey 3Osmangazi University, Faculty of Medicine, Department of Pediatrics, Division of Neonatalogy, Eskişehir, Turkey Cite this article as: Kirel B, Bozdağ Ö, Köşger P, Durmuş Aydoğdu S, Alıncak E, Tekin N. A case of Donohue syndrome “Leprechaunism” with a novel mutation in the insulin receptor gene. Turk Pediatri Ars 2017; 52: 226-30. Abstract She was diagnosed as having Donohue syndrome. Metformin and continuous nasogastric feeding were administrated. During fol- Donohue syndrome (Leprechaunism) is characterized by severe low-up, relatively good glycemic control was obtained. However, insulin resistance, hyperinsulinemia, postprandial hyperglycemia, severe hypertrophic obstructive cardiomyopathy and severe malnu- preprandial hypoglycemia, intrauterine and postnatal growth retar- trition developed. She died aged 75 days of severe heart failure and dation, dysmorphic findings, and clinical and laboratory findings pneumonia. Her insulin receptors gene analysis revealed a com- of hyperandrogenemia due to homozygous or compound heterozy- pound heterozygous mutation. One of these mutations was a p.R813 gous inactivating mutations in the insulin receptor gene. A female (c.2437C>T) mutation, which was defined previously and shown also newborn presented with lack of subcutaneous fat tissue, bilateral in her father, the other mutation was a novel p.777-790delVAAF- simian creases, hypertrichosis, especially on her face, gingival hy- PNTSSTSVPT mutation, also shown in her mother. -

Two Novel Mutations Identified in Familial Cases with Donohue
ORIGINAL ARTICLE Two novel mutations identified in familial cases with Donohue syndrome Tzipora C. Falik Zaccai1,2, Limor Kalfon1, Aharon Klar3, Mordechai Ben Elisha4, Haggit Hurvitz3, Galina Weingarten5, Emelia Chechik6, Vered Fleisher Sheffer4, Raid Haj Yahya3, Gal Meidan5, Eva Gross-Kieselstein3, Dvora Bauman7, Sylvia Hershkovitz4, Yuval Yaron8, Avi Orr-Urtreger8 & Efrat Wertheimer5 1Institute of Human Genetics, Western Galilee Medical Center, Naharia, Israel 2Faculty of Medicine in Galilee, Bar-Ilan University, Safed, Israel 3Department of Pediatrics, Bikur Cholim General Hospital, affiliated with the Hebrew University-Hadassah Medical School, Jerusalem, Israel 4Department of Neonatology, Western Galilee Medical Center, Naharia, Israel 5Department of Pathology, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel 6Sherutei Briut Clalit, Western Galilee District, Israel 7Department of Obstetrics and Gynecology, Bikur Cholim General Hospital, Jerusalem, Israel 8Genetics Institute, Tel Aviv Sourasky Medical Center, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel Keywords Abstract Cardiomyopathy, Donohue syndrome, genotype–phenotype, insulin receptor. Donohue syndrome (DS) is a rare and lethal autosomal recessive disease caused by mutations in the insulin receptor (INSR) gene, manifesting marked insulin Correspondence resistance, severe growth retardation, hypertrichosis, and characteristic dysmor- Tzipora C. Falik-Zaccai, Institute of Human phic features. We report the clinical, molecular, and biochemical characterization Genetics, Western Galilee Medical Center – of three new patients with DS, and address genotype–phenotype issues playing a Naharia, P.O. Box 21, Naharia 22100. Israel. role in the pathophysiology of DS. A female infant born to first-degree cousins Tel: 972-50-7887-941; Fax: 972-4-9107553; Muslim Arab parents and two brothers born to first-degree cousins Druze parents E-mail: [email protected] presented classical features of DS with hypertrophic cardiomyopathy and died in Funding Information infancy. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Soonerstart Automatic Qualifying Syndromes and Conditions
SoonerStart Automatic Qualifying Syndromes and Conditions - Appendix O Abetalipoproteinemia Acanthocytosis (see Abetalipoproteinemia) Accutane, Fetal Effects of (see Fetal Retinoid Syndrome) Acidemia, 2-Oxoglutaric Acidemia, Glutaric I Acidemia, Isovaleric Acidemia, Methylmalonic Acidemia, Propionic Aciduria, 3-Methylglutaconic Type II Aciduria, Argininosuccinic Acoustic-Cervico-Oculo Syndrome (see Cervico-Oculo-Acoustic Syndrome) Acrocephalopolysyndactyly Type II Acrocephalosyndactyly Type I Acrodysostosis Acrofacial Dysostosis, Nager Type Adams-Oliver Syndrome (see Limb and Scalp Defects, Adams-Oliver Type) Adrenoleukodystrophy, Neonatal (see Cerebro-Hepato-Renal Syndrome) Aglossia Congenita (see Hypoglossia-Hypodactylia) Aicardi Syndrome AIDS Infection (see Fetal Acquired Immune Deficiency Syndrome) Alaninuria (see Pyruvate Dehydrogenase Deficiency) Albers-Schonberg Disease (see Osteopetrosis, Malignant Recessive) Albinism, Ocular (includes Autosomal Recessive Type) Albinism, Oculocutaneous, Brown Type (Type IV) Albinism, Oculocutaneous, Tyrosinase Negative (Type IA) Albinism, Oculocutaneous, Tyrosinase Positive (Type II) Albinism, Oculocutaneous, Yellow Mutant (Type IB) Albinism-Black Locks-Deafness Albright Hereditary Osteodystrophy (see Parathyroid Hormone Resistance) Alexander Disease Alopecia - Mental Retardation Alpers Disease Alpha 1,4 - Glucosidase Deficiency (see Glycogenosis, Type IIA) Alpha-L-Fucosidase Deficiency (see Fucosidosis) Alport Syndrome (see Nephritis-Deafness, Hereditary Type) Amaurosis (see Blindness) Amaurosis -

Unravelling the Link Between Insulin Resistance and Androgen Excess
Unravelling the link between insulin resistance and androgen excess by Michael O’Reilly A thesis submitted to The University of Birmingham for the degree of DOCTOR OF PHILOSOPHY School of Clinical and Experimental Medicine College of Medical and Dental Sciences The University of Birmingham August 2015 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. Abstract Abstract Insulin resistance and androgen excess are the cardinal phenotypic features of polycystic ovary syndrome (PCOS). The severity of hyperandrogenism and metabolic dysfunction in PCOS are closely correlated. Aldoketoreductase type 1C3 (AKR1C3) is an important source of androgen generation in human adipose tissue, and may represent a link between androgen metabolism and metabolic disease in PCOS. We performed integrated in vitro studies using a human preadipocyte cell line and primary cultures of human adipocytes, coupled with in vivo deep phenotyping of PCOS women and age- and BMI-matched controls. We have shown that insulin upregulates AKR1C3 activity in primary female subcutaneous adipocytes. AKR1C3 mRNA expression increased with obesity. Androgens were found to increase lipid accumulation in human adipocytes. In clinical studies, androgen exposure induced relative suppression of adipose lipolysis in PCOS women, supporting a role for androgens in lipid accumulation. -

Carrier Screening Panel
Advancing Non-Invasive Healthcare Cell3TM Target: Carrier Screening Panel Detects 448 childhood recessive disorders DISORDER GENE DISEASE TYPE 2-Methylbutryl-CoA dehydrogenase deficiency ACADSB Metabolic 3-Hydroxy-3-methylglutaryl- CoA lyase deficiency HMGCL Metabolic 3-Hydroxyacyl- CoA dehydrogenase deficiency HADH Metabolic 3-Methylcrotonyl- CoA carboxylase 2 deficiency MCCC2 Metabolic 3-Methylglutaconic aciduria, i AUH Metabolic 3-Methylglutaconic aciduria, iii OPA3 Metabolic 3-Methylglutaconic aciduria, v DNAJC19 Metabolic α -Thalassemia/mental retardation syndrome, nondeletion, XLR ATRX ATRX Hematologic ABCD syndrome EDNRB Cutaneous Abetalipoproteinemia; ABL MTTP Metabolic Achalasia-Addisonianism-Alacrima syndrome; AAA AAAS Endocrine Achondrogenesis, Ib; ACG1b SLC26A2 Skeletal Achromatopsia 3; ACHM3 CNGB3 Ocular Acrocallosal syndrome; ACLS GLI3 Developmental Acyl-CoA dehydrogenase family, member 9, deficiency of ACAD9 Metabolic Acyl-CoA dehydrogenase, long-chain, deficiency of ACADL Metabolic Acyl-CoA dehydrogenase, medium-chain, deficiency of ACADM Metabolic Acyl-CoA dehydrogenase, short-chain, deficiency of ACADS Metabolic Acyl-CoA dehydrogenase, very long-chain, deficiency of ACADVL Metabolic Adrenal hyperplasia, congenital, due to 21-hydroxylase deficiency CYP21A2 Endocrine Adrenoleukodystrophy; ALD ABCD1 Neurological Afibrinogenemia, congenital FGA Hematologic Afibrinogenemia, congenital FGB Hematologic Afibrinogenemia, congenital FGG Hematologic Agammaglobulinemia, XLR, XLA BTK Immunodeficiency Agenesis of the corpus callosum -

Ocular Manifestations of Inherited Diseases Maya Eibschitz-Tsimhoni
10 Ocular Manifestations of Inherited Diseases Maya Eibschitz-Tsimhoni ecognizing an ocular abnormality may be the first step in Ridentifying an inherited condition or syndrome. Identifying an inherited condition may corroborate a presumptive diagno- sis, guide subsequent management, provide valuable prognostic information for the patient, and determine if genetic counseling is needed. Syndromes with prominent ocular findings are listed in Table 10-1, along with their alternative names. By no means is this a complete listing. Two-hundred and thirty-five of approxi- mately 1900 syndromes associated with ocular or periocular manifestations (both inherited and noninherited) identified in the medical literature were chosen for this chapter. These syn- dromes were selected on the basis of their frequency, the char- acteristic or unique systemic or ocular findings present, as well as their recognition within the medical literature. The boldfaced terms are discussed further in Table 10-2. Table 10-2 provides a brief overview of the common ocular and systemic findings for these syndromes. The table is organ- ized alphabetically; the boldface name of a syndrome is followed by a common alternative name when appropriate. Next, the Online Mendelian Inheritance in Man (OMIM™) index num- ber is listed. By accessing the OMIM™ website maintained by the National Center for Biotechnology Information at http://www.ncbi.nlm.nih.gov, the reader can supplement the material in the chapter with the latest research available on that syndrome. A MIM number without a prefix means that the mode of inheritance has not been proven. The prefix (*) in front of a MIM number means that the phenotype determined by the gene at a given locus is separate from those represented by other 526 chapter 10: ocular manifestations of inherited diseases 527 asterisked entries and that the mode of inheritance of the phe- notype has been proven. -
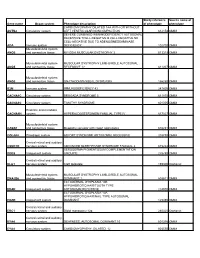
Gene Name Organ System Phenotype Description Study Reference Of
Study reference Source name of Gene name Organ system Phenotype description of phenotype phenotype CARDIOMYOPATHY DILATED 1AA WITH OR WITHOUT ACTN2 Circulatory system LEFT VENTRICULAR NONCOMPACTION 612158 OMIM SEVERE COMBINED IMMUNODEFICIENCY AUTOSOMAL RECESSIVE TCELL-NEGATIVE B CELL-NEGATIVE NK CELL-NEGATIVE DUE TO ADENOSINEDEAMINASE ADA Immune system DEFICIENCY 102700 OMIM Musculoskeletal system ANO5 and connective tissue MIYOSHI MUSCULAR DYSTROPHY 3 613319 OMIM Musculoskeletal system MUSCULAR DYSTROPHY LIMB-GIRDLE AUTOSOMAL ANO5 and connective tissue RECESSIVE 12 611307 OMIM Musculoskeletal system ANO5 and connective tissue GNATHODIAPHYSEAL DYSPLASIA 166260 OMIM B2M Immune system IMMUNODEFICIENCY 43 241600 OMIM CACNA1C Circulatory system BRUGADA SYNDROME 3 611875 OMIM CACNA1C Circulatory system TIMOTHY SYNDROME 601005 OMIM Endcrine and metabolic CACNA1H system HYPERALDOSTERONISM FAMILIAL TYPE IV 617027 OMIM Musculoskeletal system CASQ1 and connective tissue Myopathy vacuolar with casq1 aggregates 616231 OMIM COL4A4 Pleiotropic system ALPORT SYNDROME AUTOSOMAL RECESSIVE 203780 OMIM Central retinal and auditory CSNK1D nervous system ADVANCED SLEEP PHASE SYNDROME FAMILIAL 2 615224 OMIM XERODERMA PIGMENTOSUM COMPLEMENTATION DDB2 Integument system GROUP E 278740 OMIM Central retinal and auditory DLG1 nervous system Cleft lip/palate 199306 Orphanet Musculoskeletal system MUSCULAR DYSTROPHY LIMB-GIRDLE AUTOSOMAL DNAJB6 and connective tissue DOMINANT 1 603511 OMIM ECTODERMAL DYSPLASIA 10B HYPOHIDROTIC/HAIR/TOOTH TYPE EDAR Integument system -

Leprechaunism: an Inherited Defect in a High-Affinity Insulin Receptor
Am J Hum Genet 37:73 -88, 1985 Leprechaunism: An Inherited Defect in a High-Affinity Insulin Receptor L. J. ELSAS,1 F. ENDO,1 E. STRUMLAUF,1 J. ELDERS,2 AND J. H. PRIEST1 SUMMARY We examined in vivo oral glucose tolerance tests and in vitro insulin binding, cellular response, and insulin-receptor structure of fibroblasts cultured from the skin of a patient with leprechaun syndrome and her parents. In response to oral glucose, the proband exhibited marked hy- perinsulinism (maximum plasma insulin = 4,120 iiU/ml), the father had mild hyperinsulinism (maximum plasma insulin = 240 [±U/ml), and the mother was normal. [1251]insulin binding to monolayers of intact fibroblasts demonstrated complex kinetics that were interpreted using a two-receptor model. Normal high-affinity binding had an apparent KA of 1.6 x 1010/molar with 1,100 sites/cell. The proposed low-affinity state receptor had an apparent KA of 6.8 x 107/molar with approximately 30,000 sites/cell. Insulin binding to the proband's cells had no high- affinity binding but had normal low-affinity binding. Cells from the mother had 60%, and cells from the father, 2%, of control insulin binding to the high-affinity receptor, but normal, low-affinity site binding. Two different, insulin-stimulable responses were evaluated under experimental conditions identical with those used for insulin binding. Insulin stim- ulation of 2-methylaminoisobutyric acid uptake occurred with half- maximal responses between 25 and 50 ng/ml insulin. This response was similar in cells from controls and the patient. By contrast, the uptake and phosphorylation of 2-deoxy-D-glucose was stimulated at half-maximal insulin concentrations between 1 and 10 ng/ml in control cells but was not significantly increased in the proband's cells until 1,000 ng/ml con- centrations of insulin were attained. -

Severe Insulin Resistance Syndromes
Severe insulin resistance syndromes Angeliki M. Angelidi, … , Andreas Filippaios, Christos S. Mantzoros J Clin Invest. 2021;131(4):e142245. https://doi.org/10.1172/JCI142245. Review Series Severe insulin resistance syndromes are a heterogeneous group of rare disorders characterized by profound insulin resistance, substantial metabolic abnormalities, and a variety of clinical manifestations and complications. The etiology of these syndromes may be hereditary or acquired, due to defects in insulin potency and action, cellular responsiveness to insulin, and/or aberrations in adipose tissue function or development. Over the past decades, advances in medical technology, particularly in genomic technologies and genetic analyses, have provided insights into the underlying pathophysiological pathways and facilitated the more precise identification of several of these conditions. However, the exact cellular and molecular mechanisms of insulin resistance have not yet been fully elucidated for all syndromes. Moreover, in clinical practice, many of the syndromes are often misdiagnosed or underdiagnosed. The majority of these disorders associate with an increased risk of severe complications and mortality; thus, early identification and personalized clinical management are of the essence. This Review aims to categorize severe insulin resistance syndromes by disease process, including insulin receptor defects, signaling defects, and lipodystrophies. We also highlight several complex syndromes and emphasize the need to identify patients, investigate underlying disease mechanisms, and develop specific treatment regimens. Find the latest version: https://jci.me/142245/pdf The Journal of Clinical Investigation REVIEW SERIES: 100TH ANNIVERSARY OF INSULIN’S DISCOVERY Series Editor: Rexford Ahima Severe insulin resistance syndromes Angeliki M. Angelidi,1 Andreas Filippaios,2 and Christos S. Mantzoros1,3 1Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts, USA.