Joubert Syndrome: a Rare Radiological Case
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sahiwal Medical College and Allied Hospitals Sahiwal Page 1 of 47
Standard Bidding Document – Purchase of equipment and machinery – Financial Year 2019-20 STANDARD BIDDING DOCUMENT FOR THE PURCHASE MEDICAL & LAB EQUIPMENT (YEAR 2019 - 20) GOVERNEMENT OF THE PUNJAB HEALTH DEPARTMENT “SAHIWAL MEDICAL COLLEGE AND ALLIED HOSPITALS SAHIWAL” Government of the Punjab, Health Department Sahiwal Medical College and Allied Hospitals Sahiwal Page 1 of 47 Standard Bidding Document – Purchase of equipment and machinery – Financial Year 2019-20 Table of Contents Instructions To Bidder .......................................................................................................... 5 General Instructions ................................................................................................................... 5 1. Content of Bidding Documents .................................................................................................. 5 2. Source of Funds .......................................................................................................................... 5 3. Eligible Bidders ............................................................................................................................ 5 4. Eligible Goods and Services ......................................................................................................... 5 5. Cost of Bidding ............................................................................................................................ 6 6. Clarification of Bidding Documents ............................................................................................ -

Download FATA Colleges Choices Form
Pre Merit List Merit No. Declaration of Preferences PMC MDCAT Roll No. MBBS/BDS(FATA/MAD SEATS) for Agency/FR. Public Sector Medical Dental Colleges/Institutions Session 2020-21 VERY IMPORTANT: 1.Write your preferences in the order you would like to be considered for admission against the name of College/Institution. 2.Preference once given shall be final and cannot be changed subsequently. Think carefully before writing. 3.Cutting / erasing / over writing is not allowed. 4.The applicant shall never be considered for a college which he/she has not written down in this list of choices. The University shall not assign a college by itself if the alternate choices are not indicated. 5.Khyber Girls Medical College, Peshawar and Fatima Jinnah Medical College, Lahore are for female students only. Male students cannot opt for this college. 6.Candidate who do not submit choices by the deadline i.e. till 02:00 pm on 17/02/2021, the placement committee shall consider his/her choices recorded previously on the already submitted online application form. Name of the Institution/College Choice No. (In Figure and Words) Signatures of the Applicant ADS=Ayub Dental Section, Abbotabad AMC=Ayub Medical College, Abbotabad BKMC=Bacha Khan Medical College, Mardan BKDS=Bacha Khan Dental Secion, Mardan. BMC=Bannu Medical College, Bannu. GKMC = Gajju Khan Medical College Swabi KGMC = Khyber Girls Medical College (For Girls only) GMC = Gomal Medical College KCD = Khyber College of Dentistry KIMS = KMU Institute of Medical Sciences, Kohat KIDS = KMU Institute of Dental Sciences, Kohat KMC = Khyber Medical College Peshawar 1 of 2 Pre Merit List Merit No. -

Interstitial Lung Disease and Depression – a Questionnaire Based Study
ORIGINAL ARTICLE Interstitial Lung Disease and Depression – A Questionnaire Based Study Muntazir Mehdi1, Muhammad Waseem2, Hafiza Swaiba Afzal3, Ahmad Zeeshan Jamil4 Muhammad Junaid Iqbal5, Maryam Rafiq6 ABSTRACT Background and Objective: Interstitial lung disease (ILD) causes depression due to its painful course in patients. The aim of this study was to determine the prevalence of depression and find the association of depression with selected clinical variables in patients with ILD. Methods: This questionnaire based cross-sectional study was done at the department of pulmonology, District head quarter hospital Sahiwal from 1st October 2019 to 31st March 2020. The questionnaire was distributed among the diagnosed cases of ILD who presented in outpatient department of DHQ Sahiwal after taking informed consent. The depression scoring was done in them according to Beck depression inventory II. Frequency distribution statistics and inferential statistics were done by using Statistical Package for Social Sciences (SPSS) version 20. P-value < 0.05 was taken as statistically significant. Results: Depression was graded into four types according to Beck depression inventory II scoring system. Depression levels of minimal, mild, severe and extremely severe were found to have frequencies of 42.90, 14.30, 31.40 and 11.4% respectively. Depression was more prevalent in females (77.14%). Sixty two percent of severely depressed had rural background. Three fourth (75%) of severely depressed patients were from lower class group. Half of the severely depressed patients were suffering from hypertension. One fourth had ischemic heart disease. Illiteracy dominated in severely depressed where 3/4th of the participants had not received any education. Our study found statistically significant result of Beck score with socioeconomic groups (P = 0.037). -

CO for Organizations: Brochure
Clinical Odyssey for Organizations Table of Contents About Medical Joyworks, LLC 2 Clinical Odyssey for Organizations 3 Features 6 Quality Assurance 9 Pricing 11 Authors 13 International Medical Board 16 Learning Module Directory 25 1 About Medical Joyworks, LLC Medical Joyworks (MJ) is a All our content is peer reviewed by our International Medical Board, an all-volunteer physician-led company, body of more than 180 board-certified specialists and specialists in training from all offering digital products over the world. and solutions to the Our Values medical sector. Over ● We reflect our sophistication by keeping 400,000 customers– things simple. ● We like to ask questions so that we can medical practitioners, learn and progress better. students, medical schools, ● We also welcome discussions in order to learn and progress better. hospitals, governments, ● Our better ideas are those supported by clear hypotheses and data. and pharmaceutical ● We believe that quality medical content must be based on research and evidence. companies–trust MJ to ● We build products designed to solve real become better physicians, problems faced by physicians. ● Our products must remain neutral and increase sales, and unbiased always. ● Our products must remain accessible to as innovate their business. many people as possible. ● We learn the most when we are engaged, Our company was founded on October and we can stay engaged by having fun. 15th, 2010 by Dr. Nayana Somaratna and Mr. Sandaruwan Gunathilake; and we have remained an entirely self-funded operation More Information from the start. This has allowed us to build For more information, please visit: Medical Joyworks on core beliefs and https://www.medicaljoyworks.com principles, all reflected in our work and in our behavior towards ourselves and others. -

The Outcome of Limited Urethral Mobilization Urethroplasty for Anterior Hypospadias
URETHRAL MOBILIZATION URETHROPLASTY The Professional Medical Journal www.theprofesional.com ORIGINAL PROF-0-4055 DOI: 10.29309/TPMJ/2020.27.1.4055 THE OUTCOME OF LIMITED URETHRAL MOBILIZATION URETHROPLASTY FOR ANTERIOR HYPOSPADIAS. Shafiq-ur-Rehman1, Yasir Makki2, Fareena Ishtiaq3, Waleeja Shamikha4, Nauman Aziz5, Saad Fazal6 1. MBBS, FCPS Assistant Professor Paeds Surgery ABSTRACT… Objectives: Hypospadias, one of the most common genital anomalies, is Sahiwal Medical College Sahiwal. characterized by an abnormal meatal opening on the ventral aspect of penis. Anterior hypospadias 2. MBBS, FCPS are the most common. Most of the surgical techniques involves the construction of neourethra Senior Registrar Paeds Surgery Sahiwal Medical College Sahiwal. with significant risk of urethrocutaneous fistula. Limited urethral mobilization technique involves 3. MBBS the advancement of native urethra. Study Design: The objective of this study was to evaluate WMO the outcome of Limited Urethral Mobilization Urethroplasty for Anterior Hypospadias. Setting: Basic Health Unit Dhakranwali The study was conducted in the Department of Paediatric Surgery, DHQ teaching Hospital Kharian. 4. MBBS Sahiwal. Period: From January 2016 to December 2018. Material & Methods: A total number of Post Graduate Trainee 187 patients were included in this study. Limited urethral mobilization technique was used in all SIMS / Services Hospital Lahore. patients. Minimum age was 2.5 Years and maximum age was 12 years. Cosmetic appearance, 5. MBBS, M.Phil Assistant Professor functional outcome and complication rate were assessed. Results: Wound infection developed Physiology Department in 3.20 %( n=6) patients. Complete glanular disruption was seen in 2.13 %( n=4) patients. Sahiwal Medical College Sahiwal. Partial disruption of glans with meatal retraction was observed in 1.60 %( n=3) patients. -

Vol 18 Issue 02.Cdr
PATRON Prof. Arif Tajammal JAIMC Principal Allama Iqbal Medical College/ The Journal of Allama Iqbal Medical College Jinnah Hospital April - June 2020, Volume 18, Issue 02 CHIEF EDITOR Dr. Rubina Aslam Editorial i Head of Department of Psychiatry The Lady with the Lamp iii Muhamamd Imran ASSOCIATE EDITORS Muhammad Naeem Original Articles Sana Iftikhar i Mehwish Akhtar Comparison of Infection Rates Among Metacarpal Fracture Patients 162 Sabeen Irshad Managed Using K-wire with Those Managed Using Miniplate MANAGING EDITOR Muhammad Ali, Muhammad Iqbal, Yasir Iqbal, Rashid Hussain, Amjid Ali, Ali Muhammad Imran Jarrar Abbas Endoscopic Types of Gastritides and Associations of Acute Non-Erosive 167 STATISTICAL EDITORS Gastritis Mamoon Akbar Qureshi Asim Saleem, Imran Anwar Khan, Sadaf Yousaf, Touseef Anwar, Attique Abou Zarabia Pervaiz Butt Bakr, Aftab Mohsin DESIGNING & COMPOSING Distribution and Associations of Gastric Vascular Ectasias Among Patients 172 Talal Publishers Suffering Liver Cirrhosis Asim Saleem, Imran Anwar Khan, Sadaf Yousaf , Touseef Anwar, Attique Abou INTERNATIONAL ADVISORY BOARD Bakr, Aftab Mohsin Shoaib Khan (Finland) Saad Usmani (USA) Internal Hemorrhoids and Sigmoidoscopy-Assisted Rubber Band Ligation 177 Imran Anwar Khan, Asim Saleem, Sadaf Yousaf , Touseef Anwar, Attique Abou Bilal Ayub (USA) Bakr, Aftab Mohsin Adnan Agha (Saudi Arabia) Shahzad Khalid (Saudi Arabia) Clinical Audit for Adequacy if Clinical Information on CT Request Forms 181 Zeeshan Tariq (USA) Sarah Aleemi, Sidra Ali, Safia Malik Umar Farooq (USA) -

Pakistan Physiological Society PPS E-Newsletter Page # 1
Pakistan Physiological Society The Registered Society of Physiologists of Pakistan Pakistan Physiological Society PPS e-Newsletter Page # 1 Biannual Volume. 02 The Registered Society of Physiologists of Pakistan Issue. 01 PPSe-Newsletter Dear Readers, EDITORIAL Assalam-o-Alaikum. Patron Prof. Dr. Ali M. Soomro I am thankful to all the members of PPS for your kind appreciations on first issue of PPS e-Newsletter. The publication committee tried well to Editor produce second issue on time however it is now in front of you. Prof. Dr. Navaid Kazi I am really grateful to members who actively shared information regarding Associate Editors the events organized at their institutions, details are given in in this issue. Prof. Dr. Tehseen Iqbal As we did not receive complete reports from most of the institutions so I Dr. Arsalan Uqaili request the reader members in general and the councilors in particular for Members of sending complete information immediately after every activity performed Editorial Board under the flag of PPS. (2014-2017) Dr. FauziaAitazar Dr. JamshedWarsi Best regards, Dr. Noman Rashid Dr. Salman Shah Dr. SamreenBugti Dr. Khurram Prof. Dr. Navaid Kazi Editor E-Newsletter PPS Reviewers [email protected] Dr. Taseer Ali Khan Dr. ArsalanUqaili Page # 2 Biannual Volume. 02 The Registered Society of Physiologists of Pakistan Issue. 01 PPSe-Newsletter Message from PPS President (2016-2018) Prof. Dr. Zafar Hussain Tanveer Address of President PPS 2016-18 Prof. Dr. Zafar H Tanveer It is my proud privilege to be elected as President of Pakistan Physiological Society for 2016-18, which is the official, registered society of physiologists of Pakistan. -

College Inspection Report
PAKISTAN MEDICAL COMMISSION LAST RECOGNIZED INSPECTION GRADES OF PRIVATE MEDICAL COLLEGES Sr. Date of Previous Name of Institute City Grade No. Inspection 1. Abbottabad International Medical College Abbottabad. 17-12-2019 A 2. Abwa Medical College Faisalabad 20-11-2018 B 3. Aga Khan University Medical College Karachi. 05-08-2019 A+ 4. Akhtar Saeed Medical & Dental College Lahore. 26-08-2019 A 5. Al Aleem Medical College Lahore 29-08-2019 B 6. Al-Nafees Medical College Islamabad. 30-12-2015 C 7. Al-Tibri Medical College Karachi. 08-11-2013 B 8. Amna Inayat Medical College Lahore. 28-09-2017 C 9. Avicenna Medical College Lahore. 28-08-2019 A 10. Aziz Fatimah Medical & Dental College Faisalabad. 21-03-2018 C 11. Azra Naheed Medical College Lahore. 30-04-2015 B 12. Bahria University Medical College Karachi. 07-08-2019 B 13. Bakhtawar Amin Medical & Dental College Multan 20-02-2016 A 14. Baqai Medical College Karachi. 19-12-2018 F 15. Central Parks Medical College Lahore. 10-02-2015 C 16. CMH Institute of Medical Sciences Bahawalpur Bahawalpur 22-08-2019 C 17. CMH Kharian Medical College Kharian Cantt. 31-07-2019 B 18. CMH Lahore Medical College Lahore Cantt. 30-08-2019 A+ 19. CMH Multan Institute of Medical Sciences CIMS Multan Cantt 20-08-2019 A 20. Continental Medical College Lahore. 16-10-2018 C GRADING CRITERIA: 92.5% or above = A+, 85% or above = A, 77.5% or above = B, 70% or above = C, 69.9% or lower = F Sr. Date of Previous Name of Institute City Grade No. -

Distribution of Mbbs/Bds Seats
DISTRIBUTION OF MBBS/BDS SEATS MBBS SEATS IN MEDICAL INSTITUTIONS OF THE PUNJAB FJMU Sr. Category KEMU AIMC NMU (only SIMS FMU RMU QAMC SZMC SMC NSMC SLMC GMC GKMC KMSMC AMC SKZ Total No. MDC females) 1. Open Merit 302 301 280 206 191 287 297 273 126 79 90 100 100 100 100 100 90 3022 2. Reserved i. Disabled students 2 2 2 2 2 2 2 2 2 1 - - - - - - - 19 ii. Cholistan** - - - - - - - - 1 - - - - - - - - 01 iii. Underdeveloped districts *** - - 18 - - 10 - 6 15 - - - - - - - - 49 iv. FATA **** 1 1 1 1 - 1 1 1 - - - - - - - - - 07 Federal Government Share in v. - - - 47 - - - - - - - - - - - - - 47 FJMU* vi. Azad Jammu & Kashmir 1 1 4 13 2 4 5 4 2 - - - - - - - - 36 vii. Northern Areas(Gilgit-Baltistan) 8 8 1 4 3 2 9 18 2 - - - - - - - - 55 Foreign Students/Dual Nationality Holders under Pakistan Technical viii. 5 5 10 21 - 10 10 11 - - - - - - - - - 72 Assistance Programme***** (PTAP) Children of Overseas Pakistanis ix. and Dual Nationality Holders of 4 4 4 4 2 4 4 4 2 20 10 - - - - - 10 72 Pakistani Origin Reciprocal Seats x. - 1 3 - - - - 1 - - - - - - - - - 05 (for candidates of other provinces) xi. Goodwill Seats for Baluchistan 2 2 2 2 - 5 2 5 - - - - - - - - - 20 Grand Total 325 325 325 300 200 325 330 325 150 100 100 100 100 100 100 100 100 3405 Sr. Category DCD NID DSFMU Total No. 1. Open Merit 71 54 50 175 2. Reserved i. Disabled students 1 - - 1 Underdeveloped ii. 6 6 - 12 districts *** iii. FATA **** 1 2 - 3 Azad Jammu & iv. 3 - - 3 Kashmir Northern Areas(Gilgit- v. -
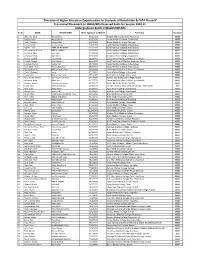
Balochistan & FATA Phase-II" Provisional Placement on MBBS/BDS Reserved Seats for Session 2020-21 Undergraduate Batch-V (BALOCHISTAN)
"Provision of Higher Education Opportunities for Students of Balochistan & FATA Phase-II" Provisional Placement on MBBS/BDS Reserved Seats for Session 2020-21 Undergraduate Batch-V (BALOCHISTAN) Sr. No NAME FATHER NAME Merit Aggregate w/MDCAT Placement Discipline 1 Wasiq Ali Tariq Tariq Bashir 93.627273 Khyber Medical College, Peshawar. MBBS 2 Rimsha Khan Nasrullah Jan 93.613636 Army Medical College, Rawalpindi. MBBS 3 Abdul Qayyum Naeem khan 93.536364 Khyber Medical College, Peshawar. MBBS 4 Asmat Ullah shams ul haq 92.845455 Ayub Medical College, Abbottabad. MBBS 5 Najeeb Ullah AMIR MUHAMMAD 92.272727 Ayub Medical College, Abbottabad. MBBS 6 Muhammad Ibrahim ABDUL JABBAR 91.650000 Ayub Medical College, Abbottabad. MBBS 7 Mohabat Khan Sado khan 91.013636 Ayub Medical College, Abbottabad. MBBS 8 Arfa Ayaz Khan Ayaz Khan 90.581818 Army Medical College, Rawalpindi. MBBS 9 Ayesha Baloch Ejaz Ahmed 90.145455 Gujranwala Medical College, Gujranwala. MBBS 10 Farooq Ahmed Altaf Hussain 89.022727 KUST Institute of Medical Sciences, Kohat. MBBS 11 Sundeep Kumar Vadoo Mal 88.740909 Ayub Medical College, Abbottabad. MBBS 12 Sana Ullah Khan Raz Muhammad 88.254545 Ayub Medical College, Abbottabad. MBBS 13 Muhammad Ali Muhammad Safdar 88.181818 Gujranwala Medical College, Gujranwala. MBBS 14 Abdul Waheed abdul 87.159091 Ayub Medical College, Abbottabad. MBBS 15 Tariq Iqbal Muhammad iqbal 85.518182 Ayub Medical College, Abbottabad. MBBS 16 Syed Haider Ali Shah Syed Asghar Ali Shah 84.440909 Nawaz Sharif Medical College, Gujrat. MBBS 17 Muheem Khan Mian dad 84.277273 Gujranwala Medical College, Gujranwala. MBBS 18 Alveena Azeem Muhammad Azeem 83.704545 Bolan Medical College, Quetta. -

Federal Capital, Islamabad Punjab Province
APPROVED PUBLIC SECTOR UNIVERSITIES / COLLEGES & THEIR CAMPUSES* Federal Capital, Islamabad Sr. # Universities / Colleges Designated Branches 1 Air University, Islamabad. Foreign Office Branch Islamabad 2 Bahria University, Islamabad. Foreign Office Branch Islamabad 3 COMSATS Institute of Information Technology, Islamabad. Foreign Office Branch Islamabad 4 Federal Urdu University of Arts, Sci. & Tech., Islamabad. Foreign Office Branch Islamabad 5 International Islamic University, Islamabad Foreign Office Branch Islamabad 6 National University of Medical Sciences, Islamabad Foreign Office Branch Islamabad 7 National University of Modern Languages, Islamabad. Foreign Office Branch Islamabad 8 National University of Science & Technology, Islamabad Foreign Office Branch Islamabad 9 National Defence University, Islamabad Foreign Office Branch Islamabad 10 Pakistan Institute of Engineering & Applied Sciences, Islamabad Foreign Office Branch Islamabad 11 Pakistan Institute of Development Economics (PIDE) Islamabad Foreign Office Branch Islamabad 12 Quaid-e-Azam University, Islamabad Foreign Office Branch Islamabad 13 Institute of Space Technology, Islamabad. Foreign Office Branch Islamabad 14 Shaheed Zulfiqar Ali Bhutto Medical University, Islamabad Foreign Office Branch Islamabad Punjab Province 1 Allama Iqbal Medical College, Lahore Main Branch Lahore. 2 Fatima Jinnah Medical College for Women, Lahore Main Branch Lahore. 3 Government College University, Lahore Main Branch Lahore. 4 King Edward Medical College, Lahore Main Branch Lahore. 5 Kinnaird College for Women, Lahore Main Branch Lahore. 6 Lahore College for Women University, Lahore. Main Branch Lahore. 7 National College of Arts, Lahore. Main Branch Lahore. 8 University of Education, Lahore. Main Branch Lahore. 9 University of Health Sciences, Lahore Main Branch Lahore. 10 University of Veterinary and Animal Sciences, Lahore. Main Branch Lahore. Sr. # Universities / Colleges Designated Branches 11 Virtual University of Pakistan, Lahore. -

Internal Medicine Residency Program at Orange Park Medical Center Is Part of the HCA Healthcare Graduate Medical Education Network
Internal Medicine RESIDENCY PROGRAM AT Orange Park Medical Center Welcome to the AboutInternal HCA Medicine Healthcare Residency Program at Orange Park Medical Center The Internal Medicine Residency Program at Orange Park Medical Center is part of the HCA Healthcare Graduate Medical Education network. HCA Healthcare is the nation’s leading provider of quality, patient-centered care. We are also the leader in graduate medical education, all brought together by a single mission: Above all else, we are committed to the care and improvement of human life. ACGME ID: 1401100937 Tracks & Positions Tracks NRMP # Available Positions Categorical 1771140C0 13 Salary PGY1 PGY2 PGY3 $52,934 $54,506 $56,212 Thank you for your interest in the Mercer University School of Medicine/ Orange Park Medical Center (OPMC) Internal Medicine Residency Program. Our IM Categorical residency program is an ACGME accredited, three-year training with 39 positions. We strive to help our residents reach their maximum potential and achieve their long- term goals; including passing their ABIM Board Exam, entering into independent practice or joining their desired fellowships in our own intuition or other well know universities. OPMC provides our residents with a state of the art 317- bed community teaching hospital with multidisciplinary services including level II trauma, labor and delivery, psychiatry, rehabilitation, adult and neonatal intensive care units. Our continuity clinic located adjacent to the hospital and at Palms Medical group is a Federally Qualified Health Care (FQHC) center. This non-for-profit organization enhances our community healthcare by providing afordable primary care and preventative services while enhancing resident education and exposure to a broad spectrum of patients.