Synthesis of Oxazoline and Oxazole Derivatives by Hypervalent- Iodine-Mediated Oxidative Cycloaddition Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Integrated Flow and Batch-Based Approach for the Synthesis of O-Methyl Siphonazole Amarcus Combined Flow and Batch Synthesis of O-Methyl Siphonazole Baumann, Ian R
LETTER 1375 An Integrated Flow and Batch-Based Approach for the Synthesis of O-Methyl Siphonazole AMarcus Combined Flow and Batch Synthesis of O-Methyl Siphonazole Baumann, Ian R. Baxendale, Malte Brasholz, John J. Hayward, Steven V. Ley, Nikzad Nikbin Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK E-mail: [email protected] Received 21 February 2011 has transformed flow chemistry from an academic interest Abstract: The bisoxazole containing natural product O-methyl si- phonazole was assembled using a suite of microreactors via a flow- to a valuable enabling technology that overcomes several based approach in concert with traditional batch methods. The use of the bottlenecks traditionally faced by synthetic chem- 6 of a toolbox of solid-supported scavengers and reagents to aid puri- ists. As such the generation and immediate use of hazard- fication afforded the natural product in a total of nine steps. ous, toxic, or unstable intermediates7 has been reported as Key words: oxazoles, Claisen condensation, flow chemistry, well as the possibility to more reliably perform reaction microreactors, solid-supported reagents scale-up.8 Furthermore, flow reactors can be transformed into automated platforms by the addition of liquid han- dling and fraction collector modules expanding the work- As part of our ongoing interest in oxazole-containing nat- ing capabilities of the units and allowing for 24/7 working 9 ural products,1 we have devised a synthetic route to the regimes. Finally, individual reactions can be telescoped bisoxazole alkaloid O-methyl siphonazole (2), which was into one continuous flow sequence, thus avoiding the iter- discovered by König and co-workers in 2006, along with ative isolation, purification, and reprocessing of interme- 10 the C-21-demethylated parent compound siphonazole (1, diates. -

Recent Syntheses of Steroidal Oxazoles, Oxazolines and Oxazolidines
A Platinum Open Access Journal Review for Organic Chemistry Free to Authors and Readers DOAJ Seal Arkivoc 2021, part i, 471-490 Recent syntheses of steroidal oxazoles, oxazolines and oxazolidines Besma Bendif,a,b Malika Ibrahim-Ouali,*a and Frédéric Dumur c aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, F-13397 Marseille, France bLaboratoire de Chimie Appliquée, Faculté des Sciences, Université du 08 mai 1945 Guelma, Algeria cAix Marseille Univ, CNRS, ICR, UMR 72 73, F-13397 Marseille, France Email: [email protected] Received 03-15-2021 Accepted 04-11-2021 Published on line 05-08-2021 Abstract It was found that the introduction of heterocycles to steroids often leads in a change of their physiological activity and the appearance of new interesting biological precursors. Recent developments in the syntheses of steroidal oxazoles, oxazolines, and oxazolidines are described herein. The biological activities of those steroidal derivatives for which data are available are given. Keywords: Steroids, oxazoles, oxazolines, oxazolidines DOI: https://doi.org/10.24820/ark.5550190.p011.512 Page 471 ©AUTHOR(S) Arkivoc 2021, i, 471-490 Bendif, B. et al. Table of Contents 1. Introduction 2. Synthesis of Steroidal Oxazoles 3. Synthesis of Steroidal Oxazolines 4. Synthesis of Steroidal Oxazolidines 5. Conclusions Acknowledgements References 1. Introduction Steroids constitute an extensive and important class of biologically active polycyclic compounds that are widely used for therapeutic purposes.1-3 Even after decades of research, the total synthesis of steroid nuclei by improved strategies continues to receive considerable attention. Numerous methods have been exploited for the total synthesis of steroids which are widely distributed in nature and which possess practical medical importance. -

Block-Poly(2-Methyl-2-Oxazoline)
ENGINEERING OF POLY(2-OXAZOLINE)S FOR A POTENTIAL USE IN BIOMEDICAL APPLICATIONS by Camille Legros A thesis presented to the University of Waterloo, Université de Liege and Université de Bordeaux in fulfillment of the thesis requirement for the degree of Doctor of Philosophy in Chemical Engineering (Nanotechnology) Waterloo, Ontario, Canada, 2015 © Camille Legros 2015 'I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public.' ii iii Abstract Résumé: Ce travail décrit d'abord l’élaboration de nanogels hydrophiles stimulables, sensibles à un changement de pH et à un environnement où les propriétés d’oxydo-réduction peuvent varier. Ils ont été synthétisés en milieu dilué, d’une part, et en émulsion inverse, d’autre part; dans les deux cas à partir d’un copolymère statistique composé d’unités 2-éthyl-2-oxazoline et éthylène imine. Ces nanogels n’ont pas montré d’interactions spécifiques avec des protéines telles que la BSA et se sont avérés non-toxiques in vitro. Une plateforme à base d’un copolymère POx statistique porteur de fonctions aldéhydes a par ailleurs permis d’accéder à une librairie de POx, incluant des structures greffées et réticulées. Enfin, l’auto-assemblage en solution d’un copolymère à blocs de type poly(2-methyl-oxazoline)-b-poly(2-isopropyl-2-oxazoline) (PMeOx- b-PiPrOx), a été étudié en détail. Des micelles ont été observées à des temps courts au-dessus du point trouble du PiPrOx. -

Novel Functional Poly(2-Oxazoline)S As Potential Carriers for Biomedical Applications
Technische Universität München Department Chemie WACKER-Lehrstuhl für Makromolekulare Chemie Novel Functional Poly(2-oxazoline)s as Potential Carriers for Biomedical Applications Robert Luxenhofer Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. Kai-Olaf Hinrichsen Prüfer der Dissertation: 1. Priv.-Doz. Dr. Rainer Jordan 2. Univ.-Prof. Dr. Horst Kessler 3. Univ.-Prof. Dr. Andreas Türler Die Dissertation wurde am 29.05.2007 bei der Technischen Universität München ein- gereicht und durch das Department Chemie am 26.06.2007 angenommen. In Vivo Veritas! Die vorliegende Arbeit wurde am WACKER-Lehrstuhl für Makromolekulare Chemie (zuvor Lehrstuhl für Makromolekulare Stoffe) im Department Chemie der Technischen Universität München in der Zeit von März 2004 bis Mai 2007 unter der Leitung von Herrn PD Dr. Rainer Jordan angefertigt. Insbesondere möchte ich mich bei meinem Mentor und Betreuer PD Dr. Rainer ´Ray´ Jordan sehr herzlich bedanken, dass er mir die Möglichkeit gegeben hat, an diesem Projekt zu arbeiten. Ich glaube das war die interessanteste Doktorarbeit die ich hätte finden können, selbst wenn ich gesucht hätte. Ich war sehr froh über all die Freiheit die ich hatte, wobei man immer vorbeikommen konnte, wenn man ein Problem hatte. Außerdem bin ich dankbar für die Arbeits- und Konferenzaufenthalte in New York, Flic en Flac, Budapest, San Francisco etc., die ich genießen konnte und die große Geduld mit mir. Prof. Dr.-Ing. Oskar Nuyken und Prof. Dr. Bernhard Rieger danke ich für die Auf- nahme bzw. die Möglichkeit meines Verbleibs am Lehrstuhl. -

And Enantioselective Synthesis of P-Hydroxy-A-Amino Acids by Condensation of Aldehydes and Ketones with Glycine
4252 J. Am. Chem. SOC.1985, 107, 4252-4259 mL and DMF and saturated with methylamine gas at 0 OC. The vessel mL volume) joined to the solvent distillation apparatus, waste, and was sealed and agitated for 1 day. The polymer was washed successively vacuum pump via Teflon tubing. in dioxane, ethanol, 2 N NaOH/i-PrOH (l:l), water (until eluate neu- In summary, we have shown for the first time the possibility to per- tral), ethanol, and ether. After drying in vacuo, the polymer (3.7 g/3.8 form highly efficient condensation reactions, by transferring polymer- mequiv of amino groups/l g of dry weight) was suspended in a mixture bound electrophiles (Le., active esters) via a mediator (shadchan) to of water (1.5 mL), ethanol (0.5 mL), triethylamine (7 mL), and 4- polymer-bound nucleophiles (i.e., amines). We have also shown the chloropyridine hydrochloride (4.7 g) in a glass pressure vessel, sealed and possibility of on-line monitoring which is relevant for automation. heated for 4 days at 140 OC. The polymer was washed as before, and The mediator methodology developed here is believed not to be limited unreacted amino groups were blocked by acetylation (acetic anhydride to acylation and related processes but to be expandable to other chemical in CH2C12,then base wash). The washed DMAP polymer was dried at processes that involve the creation of activated intermediates. These 150 OC in vacuo until constant weight. Incorporation of pyridine groups possibilities are currently under investigation. was determined by potentiometric chloride titration of the hydrochloride salt bound to the polymer: 2.53 mequiv/g compared to 3.15 mequiv/g Acknowledgment. -

European Patent Office Office Europeenpeen Des Brevets EP 0 674 618 B1
Europaisches Patentamt (19) European Patent Office Office europeenpeen des brevets EP 0 674 618 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) intci.6: C07C 317/32, C07C 233/22, of the grant of the patent: C07C 315/04, C07D 301/19, 09.09.1998 Bulletin 1998/37 C07D 263/14, A61K31/16 (21) Application number: 94903599.2 (86) International application number: PCT/US93/12071 (22) Date of filing: 15.12.1993 (87) International publication number: WO 94/14764 (07.07.1994 Gazette 1994/15) (54) ASYMMETRIC PROCESS FOR PREPARING FLORFENICOL, THIAMPHENICOL, CHLORAMPHENICOL AND OXAZOLINE INTERMEDIATES ASYMMETRISCHES HERSTELLUNGSVERFAHREN FUR FLORFENICOL, THIAMPHENICOL, CHLORAMPHENICOL UND OXAZOLIN-ZWISCHENPRODUKTE PROCEDE ASYMETRIQUE DE PREPARATION DE FLORFENICOL, THIAMPHENICOL, CHLORAMPHENICOL ET D'INTERMEDI AIRES OXAZOLINE (84) Designated Contracting States: (74) Representative: AT BE CH DE DK ES FR GB GR IE IT LI LU NL PT von Kreisler, Alek, Dipl.-Chem. et al SE Patentanwalte, von Kreisler-Selting-Werner, (30) Priority: 18.12.1992 US 993932 Bahnhofsvorplatz 1 (Deichmannhaus) 50667 Koln (DE) (43) Date of publication of application: 04.10.1995 Bulletin 1995/40 (56) References cited: EP-A- 0 423 705 EP-A- 0 472 790 (73) Proprietor: SCHERING CORPORATION WO-A-92/07824 US-A- 4 235 892 Kenilworth New Jersey 07033 (US) US-A- 4 876 352 US-A- 4 900 847 (72) Inventors: • CHEMICAL ABSTRACTS, vol. 107, no. 1, 06 July • WU, Guang-Zhong 1987, Columbus, Ohio, US; abstract no. 6859M, Somerville, NJ 08876 (US) JOMMI, GIANCARLO ET AL '2-Oxazolidinones • TORMOS, Wanda, I. as regioselective protection of beta-amino Elizabeth, NJ 07202 (US) alcohols in the synthesis of 2-amino-1-aryl-3-fluoro-1-propanols' page 632; column 1; & GAZZ. -

Recent Advances in the Synthesis of Oxazole-Based Molecules Via Van Leusen Oxazole Synthesis
molecules Review Recent Advances in the Synthesis of Oxazole-Based Molecules via van Leusen Oxazole Synthesis Xunan Zheng 1,2, Wei Liu 3,* and Dawei Zhang 1,* 1 College of Chemistry, Jilin University, Changchun 130012, China; [email protected] 2 College of Plant Science, Jilin University, Changchun 130062, China 3 Department of Pesticide Science, Plant Protection College, Shenyang Agricultural University, Shenyang 110866, China * Correspondence: [email protected] (W.L.); [email protected] (D.Z.); Tel.: +86-188-1775-2588 (W.L.); +86-431-8783-6471 (D.Z.) Academic Editors: Anna Carbone and Fabio Bertozzi Received: 2 March 2020; Accepted: 23 March 2020; Published: 31 March 2020 Abstract: Oxazole compounds, including one nitrogen atom and one oxygen atom in a five-membered heterocyclic ring, are present in various biological activities. Due to binding with a widespread spectrum of receptors and enzymes easily in biological systems through various non-covalent interactions, oxazole-based molecules are becoming a kind of significant heterocyclic nucleus, which have received attention from researchers globally, leading them to synthesize diverse oxazole derivatives. The van Leusen reaction, based on tosylmethylisocyanides (TosMICs), is one of the most appropriate strategies to prepare oxazole-based medicinal compounds. In this review, we summarize the recent advances of the synthesis of oxazole-containing molecules utilizing the van Leusen oxazole synthesis from 1972, aiming to look for potential oxazole-based medicinal compounds, which are valuable information for drug discovery and synthesis. Keywords: van Leusen; TosMICs; oxazole; synthesis 1. Introduction The oxazole ring, with one nitrogen atom and one oxygen atom, which are widely displayed in natural products and synthetic molecules, is known as a prime skeleton for drug discovery. -

Download Author Version (PDF)
Dalton Transactions Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/dalton Page 1 of 39 Dalton Transactions Chiral multidentate oxazoline ligands based on cyclophosphazene cores: Synthesis, characterization and complexation studies Dheeraj Kumar, Jatinder Singh and Anil J. Elias ,* ____________________________________________________________________ Chiral oxazoline based bi and hexadentate ligands built on cyclophosphazene cores have been synthesized and characterized. (NPPh 2)2[NP(m-OC 6H4C(O)OCH 3)2] (1) was prepared by the reaction of gem -(NPPh 2)2(NPCl 2) with methyl-3-hydroxy benzoate in presence of Cs 2CO 3. Compound 1 was converted to the dicarboxylic acid (NPPh 2)2[NP(m-OC 6H4C(O)OH)2] (2) Manuscript using base promoted hydrolysis with KO(t-Bu). -

Synthesis, Biodistribution and Excretion of Radiolabeled Poly(2-Alkyl-2-Oxazoline)S ⁎ ⁎ Florian C
Journal of Controlled Release 119 (2007) 291–300 www.elsevier.com/locate/jconrel Synthesis, biodistribution and excretion of radiolabeled poly(2-alkyl-2-oxazoline)s ⁎ ⁎ Florian C. Gaertner a,1, Robert Luxenhofer b,1, Birgit Blechert a, Rainer Jordan b, , Markus Essler a, a Nuklearmedizinische Klinik und Poliklinik, Klinikum rechts der Isar, Technische Universität München, Ismaninger Str. 22, 81675 München, Germany b Lehrstuhl für Makromolekulare Stoffe, Department Chemie, Technische Universität München, Lichtenbergstrasse 4, 85747 Garching, Germany Received 29 December 2006; accepted 19 February 2007 Available online 2 March 2007 Abstract Here we report on the preparation of well defined water-soluble poly(2-methyl-2-oxazoline) and poly(2-ethyl-2-oxazoline) terminally equipped with a chelator (N,N′,N″,N‴-tetraazacylododecane-1,4,7,10-tetraacetic acid (DOTA)) for radionuclide labeling. The tissue distribution and excretion of 111In-labeled poly(2-alkyl-2-oxazoline)s were studied in mice. We found that the hydrophilic polymers do not accumulate in tissues and are rapidly cleared from the blood pool, predominantly by glomerular filtration in the kidneys. In contrast only a small fraction is excreted via the hepatobiliary tract. Only minimal amounts of poly(2-alkyl-2-oxazoline)s are taken up by the reticuloendothelial system (RES). Scintigraphic studies revealed the feasibility of in vivo imaging of 111In-labeled poly(2-oxazoline)s. Since additional functionalities for targeting can readily be introduced into poly(2-oxazoline)s via functional monomer units, these compounds fulfill fundamental requirements for an application as carrier molecules in radionuclide therapy. © 2007 Elsevier B.V. All rights reserved. -

The Synthesis of Oxazole-Containing Natural Products by Thomas H. Graham BS, Virginia Tech, 1995 Submitted to the Graduate Facul
The Synthesis of Oxazole-containing Natural Products by Thomas H. Graham BS, Virginia Tech, 1995 Submitted to the Graduate Faculty of Arts and Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2006 UNIVERSITY OF PITTSBURGH FACULTY OF ARTS AND SCIENCES This dissertation was presented by Thomas H. Graham It was defended on January 5, 2006 and approved by Professor Paul E. Floreancig, Department of Chemistry Professor Kazunori Koide, Department of Chemistry Professor John S. Lazo, Department of Pharmacology Professor Peter Wipf, Department of Chemistry Dissertation Director ii ABSTRACT The Synthesis of Oxazole-containing Natural Products Thomas H. Graham, PhD University of Pittsburgh, 2006 The first section describes the synthesis of the C1’ to C11’ side chain of leucascandrolide A. The key step of the synthesis is a modified Robinson-Gabriel synthesis of the oxazole. The C1’ to C11’ side chain was constructed in 9 steps and 7% overall yield. The second section describes the synthesis of 2-alkynyl oxazoles and subsequent transformations into a variety of useful motifs. The conjugate addition of nucleophiles to 2-alkynyl oxazoles under basic conditions affords vinyl ethers, vinyl thioethers and enamines. The addition of ethanedithiol affords dithiolanes that can be transformed into ethyl thioesters and ketones. Nucleophilic additions of thiols to 2- alkynyl oxazolines affords oxazoline thioethers. Additions of halides under acidic conditions stereoselectively affords vinyl halides that can be further transformed by Sonogashira cross-coupling reactions. The third section describes the synthesis of (-)-disorazole C1 and analogs. The macrocycle was constructed with minimal protecting group manipulations, using mild esterification and Sonogashira cross-coupling reactions. -

Synthesis of a Novel Α, Ω-Diepoxy-Poly(2-Ethyl-2- Oxazoline) Oligomer
U.P.B. Sci. Bull., Series B, Vol. 78, Iss. 1, 2016 ISSN 1454-2331 SYNTHESIS OF A NOVEL α, ω-DIEPOXY-POLY(2-ETHYL-2- OXAZOLINE) OLIGOMER Alina-Maria ANGHELACHE1, Valentin Victor JERCA2, Florica Adriana JERCA3, Dumitru Mircea VULUGA4, Mircea TEODORESCU5 In the present paper, we report several methods of post-modification reactions to synthesize a new diepoxy-terminated poly(2-ethyl-2-oxazoline) oligomer, from α, ω-dihydroxy oligo 2-ethyl-2-oxazoline and epichlorohydrin. A comparison between phase transfer catalysis (PTC) and metalation method is made. Moreover, the influence of several factors affecting the epoxidation yield like: catalyst nature, base concentration, and temperature were investigated. The metalation method was very sensitive to water traces and therefore the yield was lower due to the very hygroscopic character of the poly(2-ethyl-2-oxazoline). The PTC method is more suitable for the synthesis of diepoxy oligomers and the optimal ratio between the reactants was determined. Keywords: 2-ethyl-2-oxazoline, cationic polymerization, epichlorohydrin, phase- transfer catalyst 1. Introduction Since their discovery in 1966 [1, 2], the class of 2-oxazolines has received great interest due to its versatility in enabling the preparation of materials with tailor-made properties, such as: fluorescent azo-polymers [3], light-responsive azo-polymers [4], thermo-responsive [5] polymers, and so on. The living cationic ring-opening polymerization (CROP) of 2-oxazolines provides easy and direct access to a wide variety of well-defined polymers [6, 7], in which the endgroup 1 PhD student, Department of Bioresources and Polymer Science, Faculty of Applied Chemistry and Materials Science, University POLITEHNICA of Bucharest, and Centre of Organic Chemistry “Costin D. -
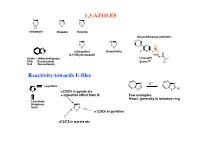
1,3-AZOLES Reactivity Towards E-Files
1,3-AZOLES N N N N S O H Imidazole Thiazole Oxazole Oxazolidineone antibiotic N HN O N O N O O 2-Oxazoline Oxazolidine F N O X (4,5-Dihydrooxazol) H N X=NH: 1H-Benzimidazole Linesolid X=O: Benzoxazole ZyvoxTM O X=S Benzotiazole Reactivity towards E-files N E+ N N ≈ pyridine X X E X ≈C3/C4 in pyrole etc + inductive effect from N Few examples React. generally in benzene ring ≈ pyrazole thiophene N furan X ≈ C2/C6 in pyridine ≈C2/C5 in pyrole etc Reaction with electrophiles on N - Protonation H H pKa H N H+ N N N H X=NH: 7.1 N X X X=S: 2.5 N N X=O: 0.8 H H O stabilized Destabilized R R N HN Taut.: N N H R= Me: ca 1 : 1 R=NO2: ca 400 : 1 Reaction with electrophiles on N R 'R R 'R R N-Alkylation N N N N N N H H R'-X R N R-X N - H+ R R R X X HN HN N May be sterically favoured Reactivity: N N N R' R' X = N-Me X=S X=O 900 : 15 : 1 R R R N N N N low react. N N N N H R' SO2Ph Base R'-X - H+ R R 'R R HN N N N N N N N H Base Et Et N + - N N PhCOCl 1) Et3O BF4 N N N O O HN N steric control Reaction with electrophiles on N N-Acylation Only rel.