39UTR Shortening Identifies High-Risk Cancers with Targeted Dysregulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Title a New Centrosomal Protein Regulates Neurogenesis By
Title A new centrosomal protein regulates neurogenesis by microtubule organization Authors: Germán Camargo Ortega1-3†, Sven Falk1,2†, Pia A. Johansson1,2†, Elise Peyre4, Sanjeeb Kumar Sahu5, Loïc Broic4, Camino De Juan Romero6, Kalina Draganova1,2, Stanislav Vinopal7, Kaviya Chinnappa1‡, Anna Gavranovic1, Tugay Karakaya1, Juliane Merl-Pham8, Arie Geerlof9, Regina Feederle10,11, Wei Shao12,13, Song-Hai Shi12,13, Stefanie M. Hauck8, Frank Bradke7, Victor Borrell6, Vijay K. Tiwari§, Wieland B. Huttner14, Michaela Wilsch- Bräuninger14, Laurent Nguyen4 and Magdalena Götz1,2,11* Affiliations: 1. Institute of Stem Cell Research, Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany. 2. Physiological Genomics, Biomedical Center, Ludwig-Maximilian University Munich, Germany. 3. Graduate School of Systemic Neurosciences, Biocenter, Ludwig-Maximilian University Munich, Germany. 4. GIGA-Neurosciences, Molecular regulation of neurogenesis, University of Liège, Belgium 5. Institute of Molecular Biology (IMB), Mainz, Germany. 6. Instituto de Neurociencias, Consejo Superior de Investigaciones Científicas and Universidad Miguel Hernández, Sant Joan d’Alacant, Spain. 7. Laboratory for Axon Growth and Regeneration, German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany. 8. Research Unit Protein Science, Helmholtz Centre Munich, German Research Center for Environmental Health, Munich, Germany. 9. Protein Expression and Purification Facility, Institute of Structural Biology, Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany. 10. Institute for Diabetes and Obesity, Monoclonal Antibody Core Facility, Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany. 11. SYNERGY, Excellence Cluster of Systems Neurology, Biomedical Center, Ludwig- Maximilian University Munich, Germany. 12. Developmental Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, USA 13. -

Functional Annotation of Exon Skipping Event in Human Pora Kim1,*,†, Mengyuan Yang1,†,Keyiya2, Weiling Zhao1 and Xiaobo Zhou1,3,4,*
D896–D907 Nucleic Acids Research, 2020, Vol. 48, Database issue Published online 23 October 2019 doi: 10.1093/nar/gkz917 ExonSkipDB: functional annotation of exon skipping event in human Pora Kim1,*,†, Mengyuan Yang1,†,KeYiya2, Weiling Zhao1 and Xiaobo Zhou1,3,4,* 1School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA, 2College of Electronics and Information Engineering, Tongji University, Shanghai, China, 3McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA and 4School of Dentistry, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA Received August 13, 2019; Revised September 21, 2019; Editorial Decision October 03, 2019; Accepted October 03, 2019 ABSTRACT been used as therapeutic targets (3–8). For example, MET has lost the binding site of E3 ubiquitin ligase CBL through Exon skipping (ES) is reported to be the most com- exon 14 skipping event (9), resulting in an enhanced expres- mon alternative splicing event due to loss of func- sion level of MET. MET amplification drives the prolifera- tional domains/sites or shifting of the open read- tion of tumor cells. Multiple tyrosine kinase inhibitors, such ing frame (ORF), leading to a variety of human dis- as crizotinib, cabozantinib and capmatinib, have been used eases and considered therapeutic targets. To date, to treat patients with MET exon 14 skipping (10). Another systematic and intensive annotations of ES events example is the dystrophin gene (DMD) in Duchenne mus- based on the skipped exon units in cancer and cular dystrophy (DMD), a progressive neuromuscular dis- normal tissues are not available. -

Bioinformatics Identi Cation of Prognostic Factors Associated With
Bioinformatics Identication of Prognostic Factors Associated with Breast Cancer Ying Wei Sichuan University https://orcid.org/0000-0001-8178-4705 Shipeng Zhang College of Pharmacy, North Sichuan Medical College Li Xiao West China School of Basic Medical Sciences and Forensic Medicine Jing Zou West China School of Basic Medical Sciences and Forensic Medicine Yingqing Fu West China School of Basic Medical Sciences and Forensic Medicine Yi Ye West China School of Basic Medical Sciences and Forensic Medicine Linchuan Liao ( [email protected] ) West China School of Basic Medical Sciences and Forensic Medicine https://orcid.org/0000-0003-3700-8471 Research Keywords: Breast cancer, Differentially expressed genes, miRNAs, Transcription factors, Bioinformatic analysis Posted Date: December 2nd, 2020 DOI: https://doi.org/10.21203/rs.3.rs-117477/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract Background: Breast cancer (BRCA) remains one of the most common forms of cancer and is the most prominent driver of cancer-related death among women. The mechanistic basis for BRCA, however, remains incompletely understood. In particular, the relationships between driver mutations and signaling pathways in BRCA are poorly characterized, making it dicult to identify reliable clinical biomarkers that can be employed in diagnostic, therapeutic, or prognostic contexts. Methods: First, we downloaded publically available BRCA datasets (GSE45827, GSE42568, and GSE61304) from the Gene Expression Omnibus (GEO) database. We then compared gene expression proles between tumor and control tissues in these datasets using Venn diagrams and the GEO2R analytical tool. We further explore the functional relevance of BRCA-associated differentially expressed genes (DEGs) via functional and pathway enrichment analyses using the DAVID tool, and we then constructed a protein-protein interaction network incorporating DEGs of interest using the Search Tool for the Retrieval of Interacting Genes (STRING) database. -

Multiple Activities of Arl1 Gtpase in the Trans-Golgi Network Chia-Jung Yu1,2 and Fang-Jen S
© 2017. Published by The Company of Biologists Ltd | Journal of Cell Science (2017) 130, 1691-1699 doi:10.1242/jcs.201319 COMMENTARY Multiple activities of Arl1 GTPase in the trans-Golgi network Chia-Jung Yu1,2 and Fang-Jen S. Lee3,4,* ABSTRACT typical features of an Arf-family GTPase, including an amphipathic ADP-ribosylation factors (Arfs) and ADP-ribosylation factor-like N-terminal helix and a consensus site for N-myristoylation (Lu et al., proteins (Arls) are highly conserved small GTPases that function 2001; Price et al., 2005). In yeast, recruitment of Arl1 to the Golgi as main regulators of vesicular trafficking and cytoskeletal complex requires a second Arf-like GTPase, Arl3 (Behnia et al., reorganization. Arl1, the first identified member of the large Arl family, 2004; Setty et al., 2003). Yeast Arl3 lacks a myristoylation site and is an important regulator of Golgi complex structure and function in is, instead, N-terminally acetylated; this modification is required for organisms ranging from yeast to mammals. Together with its effectors, its recruitment to the Golgi complex by Sys1. In mammalian cells, Arl1 has been shown to be involved in several cellular processes, ADP-ribosylation-factor-related protein 1 (Arfrp1), a mammalian including endosomal trans-Golgi network and secretory trafficking, lipid ortholog of yeast Arl3, plays a pivotal role in the recruitment of Arl1 droplet and salivary granule formation, innate immunity and neuronal to the trans-Golgi network (TGN) (Behnia et al., 2004; Panic et al., development, stress tolerance, as well as the response of the unfolded 2003b; Setty et al., 2003; Zahn et al., 2006). -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Datasheet PB1029 Anti-AEBP2 Antibody
Product datasheet Anti-AEBP2 Antibody Catalog Number: PB1029 BOSTER BIOLOGICAL TECHNOLOGY Special NO.1, International Enterprise Center, 2nd Guanshan Road, Wuhan, China Web: www.boster.com.cn Phone: +86 27 67845390 Fax: +86 27 67845390 Email: [email protected] Basic Information Product Name Anti-AEBP2 Antibody Gene Name AEBP2 Source Rabbit IgG Species Reactivity human,mouse,rat Tested Application WB,IHC-P,ICC/IF,FCM Contents 500ug/ml antibody with PBS ,0.02% NaN3 , 1mg BSA and 50% glycerol. Immunogen E.coli-derived human AEBP2 recombinant protein (Position: K424-Q517). Human AEBP2 shares 98.8% amino acid (aa) sequence identity with mouse AEBP2. Purification Immunogen affinity purified. Observed MW 54KD Dilution Ratios Western blot: 1:500-2000 Immunohistochemistry(Paraffin-embedded Section): 1:50-400 Immunocytochemistry/Immunofluorescence (ICC/IF): 1:50-400 Flow cytometry (FCM): 1-3μg/1x106 cells Storage 12 months from date of receipt,-20℃ as supplied.6 months 2 to 8℃ after reconstitution. Avoid repeated freezing and thawing Background Information Adipocyte Enhancer-Binding Protein is a zinc finger protein that in humans is encoded by the evolutionarily well-conserved gene AEBP2. This gene is mapped to 12p12.3. AEBP2 is a DNA-binding transcriptional repressor. It may regulate the migration and development of the neural crest cells through the PRC2-mediated epigenetic mechanism and is most likely a targeting protein for the mammalian PRC2 complex. Reference Anti-AEBP2 Antibody被引用在0文献中。 暂无引用 FOR RESEARCH USE ONLY. NOT FOR DIAGNOSTIC AND CLINICAL USE. 1 Product datasheet Anti-AEBP2 Antibody Catalog Number: PB1029 BOSTER BIOLOGICAL TECHNOLOGY Special NO.1, International Enterprise Center, 2nd Guanshan Road, Wuhan, China Web: www.boster.com.cn Phone: +86 27 67845390 Fax: +86 27 67845390 Email: [email protected] Selected Validation Data Figure 1. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Transcriptomic Analysis of Native Versus Cultured Human and Mouse Dorsal Root Ganglia Focused on Pharmacological Targets Short
bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia focused on pharmacological targets Short title: Comparative transcriptomics of acutely dissected versus cultured DRGs Andi Wangzhou1, Lisa A. McIlvried2, Candler Paige1, Paulino Barragan-Iglesias1, Carolyn A. Guzman1, Gregory Dussor1, Pradipta R. Ray1,#, Robert W. Gereau IV2, # and Theodore J. Price1, # 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA 2Washington University Pain Center and Department of Anesthesiology, Washington University School of Medicine # corresponding authors [email protected], [email protected] and [email protected] Funding: NIH grants T32DA007261 (LM); NS065926 and NS102161 (TJP); NS106953 and NS042595 (RWG). The authors declare no conflicts of interest Author Contributions Conceived of the Project: PRR, RWG IV and TJP Performed Experiments: AW, LAM, CP, PB-I Supervised Experiments: GD, RWG IV, TJP Analyzed Data: AW, LAM, CP, CAG, PRR Supervised Bioinformatics Analysis: PRR Drew Figures: AW, PRR Wrote and Edited Manuscript: AW, LAM, CP, GD, PRR, RWG IV, TJP All authors approved the final version of the manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Identification of BBX Proteins As Rate-Limiting Co-Factors of HY5
1 Identification of BBX proteins as rate-limiting co-factors of HY5. 2 Katharina Bursch1, Gabriela Toledo-Ortiz2, Marie Pireyre3, Miriam Lohr1, Cordula Braatz1, Henrik 3 Johansson1* 4 1. Institute of Biology/Applied Genetics, Dahlem Centre of Plant Sciences (DCPS), Freie Universität 5 Berlin, Albrecht-Thaer-Weg 6. D-14195 Berlin, Germany. 6 2. Lancaster Environment Centre, Lancaster University, Lancaster LA1 4YQ, UK 7 3. Department of Botany and Plant Biology, Section of Biology, Faculty of Sciences, University of 8 Geneva, 30 Quai E. Ansermet, 1211 Geneva 4, Switzerland 9 *E-mail [email protected] 10 Abstract 11 As a source of both energy and environmental information, monitoring the incoming light is crucial for 12 plants to optimize growth throughout development1. Concordantly, the light signalling pathways in 13 plants are highly integrated with numerous other regulatory pathways2,3. One of these signal 14 integrators is the bZIP transcription factor HY5 which holds a key role as a positive regulator of light 15 signalling in plants4,5. Although HY5 is thought to act as a DNA-binding transcriptional regulator6,7, the 16 lack of any apparent transactivation domain8 makes it unclear how HY5 is able to accomplish its many 17 functions. Here, we describe the identification of three B-box containing proteins (BBX20, 21 and 22) 18 as essential partners for HY5 dependent modulation of hypocotyl elongation, anthocyanin 19 accumulation and transcriptional regulation. The bbx202122 triple mutant mimics the phenotypes of 20 hy5 in the light and its ability to suppress the cop1 mutant phenotype in darkness. Furthermore, 84% 21 of genes that exhibit differential expression in bbx202122 are also HY5 regulated, and we provide 22 evidence that HY5 requires the B-box proteins for transcriptional regulation. -
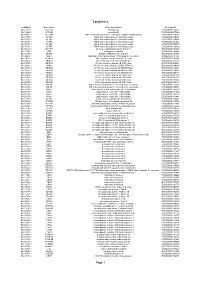
Epigenetics Page 1
Epigenetics esiRNA ID Gene Name Gene Description Ensembl ID HU-13237-1 ACTL6A actin-like 6A ENSG00000136518 HU-13925-1 ACTL6B actin-like 6B ENSG00000077080 HU-14457-1 ACTR1A ARP1 actin-related protein 1 homolog A, centractin alpha (yeast) ENSG00000138107 HU-10579-1 ACTR2 ARP2 actin-related protein 2 homolog (yeast) ENSG00000138071 HU-10837-1 ACTR3 ARP3 actin-related protein 3 homolog (yeast) ENSG00000115091 HU-09776-1 ACTR5 ARP5 actin-related protein 5 homolog (yeast) ENSG00000101442 HU-00773-1 ACTR6 ARP6 actin-related protein 6 homolog (yeast) ENSG00000075089 HU-07176-1 ACTR8 ARP8 actin-related protein 8 homolog (yeast) ENSG00000113812 HU-09411-1 AHCTF1 AT hook containing transcription factor 1 ENSG00000153207 HU-15150-1 AIRE autoimmune regulator ENSG00000160224 HU-12332-1 AKAP1 A kinase (PRKA) anchor protein 1 ENSG00000121057 HU-04065-1 ALG13 asparagine-linked glycosylation 13 homolog (S. cerevisiae) ENSG00000101901 HU-13552-1 ALKBH1 alkB, alkylation repair homolog 1 (E. coli) ENSG00000100601 HU-06662-1 ARID1A AT rich interactive domain 1A (SWI-like) ENSG00000117713 HU-12790-1 ARID1B AT rich interactive domain 1B (SWI1-like) ENSG00000049618 HU-09415-1 ARID2 AT rich interactive domain 2 (ARID, RFX-like) ENSG00000189079 HU-03890-1 ARID3A AT rich interactive domain 3A (BRIGHT-like) ENSG00000116017 HU-14677-1 ARID3B AT rich interactive domain 3B (BRIGHT-like) ENSG00000179361 HU-14203-1 ARID3C AT rich interactive domain 3C (BRIGHT-like) ENSG00000205143 HU-09104-1 ARID4A AT rich interactive domain 4A (RBP1-like) ENSG00000032219 HU-12512-1 ARID4B AT rich interactive domain 4B (RBP1-like) ENSG00000054267 HU-12520-1 ARID5A AT rich interactive domain 5A (MRF1-like) ENSG00000196843 HU-06595-1 ARID5B AT rich interactive domain 5B (MRF1-like) ENSG00000150347 HU-00556-1 ASF1A ASF1 anti-silencing function 1 homolog A (S. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

CDC2 Mediates Progestin Initiated Endometrial Stromal Cell Proliferation: a PR Signaling to Gene Expression Independently of Its Binding to Chromatin
CDC2 Mediates Progestin Initiated Endometrial Stromal Cell Proliferation: A PR Signaling to Gene Expression Independently of Its Binding to Chromatin Griselda Vallejo1, Alejandro D. La Greca1., Inti C. Tarifa-Reischle1., Ana C. Mestre-Citrinovitz1, Cecilia Ballare´ 2, Miguel Beato2,3, Patricia Saragu¨ eta1* 1 Instituto de Biologı´a y Medicina Experimental, IByME-Conicet, Buenos Aires, Argentina, 2 Centre de Regulacio´ Geno`mica, (CRG), Barcelona, Spain, 3 University Pompeu Fabra (UPF), Barcelona, Spain Abstract Although non-genomic steroid receptor pathways have been studied over the past decade, little is known about the direct gene expression changes that take place as a consequence of their activation. Progesterone controls proliferation of rat endometrial stromal cells during the peri-implantation phase of pregnancy. We showed that picomolar concentration of progestin R5020 mimics this control in UIII endometrial stromal cells via ERK1-2 and AKT activation mediated by interaction of Progesterone Receptor (PR) with Estrogen Receptor beta (ERb) and without transcriptional activity of endogenous PR and ER. Here we identify early downstream targets of cytoplasmic PR signaling and their possible role in endometrial stromal cell proliferation. Microarray analysis of global gene expression changes in UIII cells treated for 45 min with progestin identified 97 up- and 341 down-regulated genes. The most over-represented molecular functions were transcription factors and regulatory factors associated with cell proliferation and cell cycle, a large fraction of which were repressors down-regulated by hormone. Further analysis verified that progestins regulate Ccnd1, JunD, Usf1, Gfi1, Cyr61, and Cdkn1b through PR- mediated activation of ligand-free ER, ERK1-2 or AKT, in the absence of genomic PR binding.