Carbo-Deoxygenation of Biomass: the Carbonylation of Glycerol and Higher Poly-Ols to Mono-Carboxylic Acids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TOX-48: Allyl Acetate (CASRN 591-87-7), Allyl Alcohol (CASRN
National Toxicology Program Toxicity Report Series Number 48 NTP Technical Report on the Comparative Toxicity Studies of Allyl Acetate, Allyl Alcohol, and Acrolein (CAS Nos. 591-87-7, 107-18-6, and 107-02-8) Administered by Gavage to F344/N Rats and B6C3F1 Mice July 2006 National Institutes of Health Public Health Service U.S. Department of Health and Human Services FOREWORD The National Toxicology Program (NTP) is an interagency program within the Public Health Service (PHS) of the Department of Health and Human Services (HHS) and is headquartered at the National Institute of Environmental Health Sciences of the National Institutes of Health (NIEHS/NIH). Three agencies contribute resources to the program: NIEHS/NIH, the National Institute for Occupational Safety and Health of the Centers for Disease Control and Prevention (NIOSH/CDC), and the National Center for Toxicological Research of the Food and Drug Administration (NCTR/FDA). Established in 1978, the NTP is charged with coordinating toxicological testing activities, strengthening the science base in toxicology, developing and validating improved testing methods, and providing information about potentially toxic substances to health regulatory and research agencies, scientific and medical communities, and the public. The Toxicity Study Report series began in 1991. The studies described in the Toxicity Study Report series are designed and conducted to characterize and evaluate the toxicologic potential of selected substances in laboratory animals (usually two species, rats and mice). Substances selected for NTP toxicity studies are chosen primarily on the basis of human exposure, level of production, and chemical structure. The interpretive conclusions presented in the Toxicity Study Reports are based only on the results of these NTP studies. -

Acetoxylation of Olefins Over Supported Palladium Metal and Salts Catalysts*
Acetoxylation of Olefins over Supported Palladium Metal and Salts Catalysts* by Taiseki Kunugi**, HiromichiArai** and Kaoru Fujimoto** Summary: The acetoxylationof olefinsin thegaseous phase overpalladium metal and salts-active charcoalcatalysts was studiedusing a fixed bed reactor. Palladium salts adsorbedon activecharcoal are excellentcatalysts for this reactioneven in the absenceof any usual redoxreagents such as CuCl2. It was concludedthat the reoxidationof reducedpalladium is catalyzedby active charcoalin thepre- senceof oxygen. In the caseof acetoxylationof ethylenethe mainproduct was vinyl acetatefor both catalysts, but in the caseof propylene, allyl acetate,isopropenyl acetate and acetonewere mainlyproduced over palla- dium salts-activecharcoal catalysts, whereasallyl acetatewas the only principal product overpalla- dium metal-activecharcoal catalyst. The differencein theproduct distributionbetween these catalysts supposedly indicates that propylene forms a π-complex with palladium salts to give allyl acetate and isopropenylacetate whichwas hydrolyzedto form acetoneand, on the other hand, that propylene forms π-allyl complex with palladium metal to give allyl acetate. The kineticsof acetoxylationof ethylenewas studiedwith a differentialreactor for both catalysts and the reactionscheme was proposed for eachreaction system. 1 Introduction proceed by a similar mechanism2). The present authors have shown that palladium The oxidation of olefins to vinyl and allylic salts adsorbed on active charcoal are very effici- acetates has been reported recently. In the case ent catalysts for the Wacker reaction and for the of ethylene, as first reported by Moiseev et al.1) acetoxylation of olefins as well despite of the ab- the reaction is described by the following equation: sence of redox reagents such as CuCl27)~9). Ethyl- C2H4+Pd2++2OAc- ene reacts with acetic acid on palladium salts or →C2H3OAc+HOAc+Pd2 (1) palladium metal to produce vinyl acetate. -

Ep 2531496 B1
(19) TZZ ¥__T (11) EP 2 531 496 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07D 301/12 (2006.01) C07D 303/16 (2006.01) 15.03.2017 Bulletin 2017/11 (86) International application number: (21) Application number: 11701942.2 PCT/EP2011/000322 (22) Date of filing: 26.01.2011 (87) International publication number: WO 2011/095294 (11.08.2011 Gazette 2011/32) (54) MANUFACTURE OF AN EPOXYETHYL CARBOXYLATE OR GLYCIDYL CARBOXYLATE HERSTELLUNG VON EPOXYDETHYLCARBOXYLAT ODER GLYCIDYLCARBOXYLAT FABRICATION DE CARBOXYLATE ÉPOXYÉHTYL OU CARBOXYLATE GLYCIDYLIQUE (84) Designated Contracting States: (56) References cited: AL AT BE BG CH CY CZ DE DK EE ES FI FR GB EP-A1- 0 618 202 EP-A1- 2 149 569 GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO WO-A1-2005/089549 US-A- 4 423 071 PL PT RO RS SE SI SK SM TR US-A- 6 136 991 US-A1- 2003 069 439 US-A1- 2006 100 348 (30) Priority: 02.02.2010 EP 10001034 • DE VOS D E ET AL: "Epoxidation of Terminal or (43) Date of publication of application: Electron-deficient Olefins with H2O2, catalysed 12.12.2012 Bulletin 2012/50 by Mn-trimethyltriazacyclonane Complexes in the Presence of an Oxalate Buffer", (73) Proprietor: Hexion Research Belgium SA TETRAHEDRON LETTERS, ELSEVIER, 1348 Ottignies Louvain-la-Neuve (BE) AMSTERDAM, NL LNKD- DOI:10.1016/S0040-4039(98)00205-6, vol. 39, no. (72) Inventors: 20, 14 May 1998 (1998-05-14), pages 3221-3224, • MUPPA, Prasad XP004116236, ISSN: 0040-4039 NL-3196 KK Vondelingenplaat Rt (NL) • BRACE N. -

Revisiting the Deoxydehydration of Glycerol Towards Allyl Alcohol Under Continuous-Flow Conditions
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is © The Royal Society of Chemistry 2017 Supporting Information for: Revisiting the deoxydehydration of glycerol towards allyl alcohol under continuous-flow conditions Nelly Ntumba Tshibalonza and Jean-Christophe M. Monbaliu* Center for Integrated Technology and Organic Synthesis, Department of Chemistry, University of Liège, B-4000 Liège (Sart Tilman), Belgium Table of Contents 1. Continuous-flow setups .................................................................................................................3 1.1 Generalities..............................................................................................................................3 1.1.1 Pumps ..............................................................................................................................3 1.1.2 Microfluidic reactor elements..........................................................................................3 1.1.3 Thermoregulated oven.....................................................................................................5 1.1.4 In-line IR spectrometer....................................................................................................5 1.2 Detailed microfluidic setup .....................................................................................................6 1.3 Operation to steady state..........................................................................................................6 1.4.1 Starting up procedure.......................................................................................................6 -

Provisional Peer-Reviewed Toxicity Values for Allyl Alcohol (Casrn 107-18-6)
EPA/690/R-09/001F l Final 09-29-2009 Provisional Peer-Reviewed Toxicity Values for Allyl alcohol (CASRN 107-18-6) Superfund Health Risk Technical Support Center National Center for Environmental Assessment Office of Research and Development U.S. Environmental Protection Agency Cincinnati, OH 45268 Commonly Used Abbreviations BMD Benchmark Dose IRIS Integrated Risk Information System IUR inhalation unit risk LOAEL lowest-observed-adverse-effect level LOAELADJ LOAEL adjusted to continuous exposure duration LOAELHEC LOAEL adjusted for dosimetric differences across species to a human NOAEL no-observed-adverse-effect level NOAELADJ NOAEL adjusted to continuous exposure duration NOAELHEC NOAEL adjusted for dosimetric differences across species to a human NOEL no-observed-effect level OSF oral slope factor p-IUR provisional inhalation unit risk p-OSF provisional oral slope factor p-RfC provisional inhalation reference concentration p-RfD provisional oral reference dose RfC inhalation reference concentration RfD oral reference dose UF uncertainty factor UFA animal to human uncertainty factor UFC composite uncertainty factor UFD incomplete to complete database uncertainty factor UFH interhuman uncertainty factor UFL LOAEL to NOAEL uncertainty factor UFS subchronic to chronic uncertainty factor i FINAL 9-29-2009 PROVISIONAL PEER-REVIEWED TOXICITY VALUES FOR ALLYL ALCOHOL (CASRN 107-18-6) Background On December 5, 2003, the U.S. Environmental Protection Agency's (U.S. EPA) Office of Superfund Remediation and Technology Innovation (OSRTI) revised its hierarchy of human health toxicity values for Superfund risk assessments, establishing the following three tiers as the new hierarchy: 1) U.S. EPA's Integrated Risk Information System (IRIS). 2) Provisional Peer-Reviewed Toxicity Values (PPRTVs) used in U.S. -

United States Patent Office Patented June 23, 1970 2 (3) the First Gaseous Mixture Consists Essentially of 3,517,054 PROCESS for PREPARNG
3,517,054 United States Patent Office Patented June 23, 1970 2 (3) The first gaseous mixture consists essentially of 3,517,054 PROCESS FOR PREPARNG. MONOACETN about 40-60 parts by volume of propylene, 40-60 parts FROM PROPYLENE by volume of acetic acid vapor, and 5-15 parts by volume Arthur D. Ketley, Silver Spring, Md., assignor to W. R. of oxygen; Grace & Co., New York, N.Y., a corporation of (4) The catalyst is maintained at about 100-150° C.; Connecticut (5) The allyl acetate is added to an acid selected No Drawing. Filed Dec. 5, 1967, Ser. No. 688,012 from the group consisting of formic acid and acetic int. C. C07 c 67/00, 67/04 acid at a rate of about 0.5-2 moles per liter of said U.S. C. 260-491 8 Claims acid; 10 (6) The hydrogen peroxide is added to the third mix ture at a rate to provide about 100-150 grams of H2O2 ABSTRACT OF THE DISCLOSURE per mole of allyl acetate present in the third mixture; In abstract, this invention is directed to a process for and preparing monoacetin from propylene, said process com (7) The fourth mixture is maintained at about 20 prising forming allyl acetate from propylene by catalyti 5 40°C. for about 0.25-24 hours. cally reacting gaseous propylene with acetic acid vapor U.S. Pat. 3,275,680 discloses the preparation of allyl in the presence of a catalyst and gaseous oxygen, separat alcohol from propylene by a process comprising the ing the allyl acetate, and forming monoacetin by dis catalytic reaction of propylene oxygen (air), and po solving the allyl acetate in an acid selected from the tassium acetate in the presence of water. -

Polymers of Allyl Esters with Allylic Alcohols Or Propoxylated Allylic
Europaisches Patentamt (19) European Patent Office Office europeenpeen des brevets EP 0 703 250 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) intci.e: C08F 218/12, C08F 216/08 of the grant of the patent: 07.01.1999 Bulletin 1999/01 (21) Application number: 95306572.9 (22) Date of filing: 18.09.1995 (54) Polymers of ally I esters with ally lie alcohols or propoxylated ally lie alcohols Polymere aus Allylestern mit Allylalkohol oder Allyl-Alkohol-Propoxylate Polymeres d'esters allyliques et d'alcool allylique ou d'alcool allylique propoxyle (84) Designated Contracting States: (74) Representative: Cropp, John Anthony David et al BE DE ES FR GB IT NL MATHYS& SQUIRE 100 Grays Inn Road (30) Priority: 21.09.1994 US 309699 London, WC1X8AL (GB) 15.05.1995 US 440865 (56) References cited: (43) Date of publication of application: DE-A- 4 135 399 FR-A-2 018 263 27.03.1996 Bulletin 1996/13 US-A- 2 995 535 US-A- 3 876 588 (73) Proprietor: ARCO Chemical Technology, L.P. Remarks: Greenville, Delaware 19807 (US) The file contains technical information submitted after the application was filed and not included in this (72) Inventor: Guo, Shao-Hua specification West Goshen, PA 19380 (US) DO o CM CO o Is- Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice the Patent Office of the Notice of shall be filed in o to European opposition to European patent granted. opposition a written reasoned statement. -

Allyl Acetate CAS # 591-87-7
SUMMARY OF DATA FOR CHEMICAL SELECTION Allyl Acetate CAS # 591-87-7 and Allyl Alcohol CAS # 107-18-6 (Prepared for NCI by Technical Resources, Inc. under contract - (8/91: revised 2/93)) NTP NOMINATION HISTORY AND REVIEW Nomination History • Nomination Source: National Cancer Institute Recommendations: • Metabolism • Carcinogenicity Rationale/Remarks: • Allyl acetate and allyl alcohol were nominated as a pair of chemicals • High production • Potential for high occupational and consumer exposure based on its extensive use in organic synthesis, agriculture and flavoring agents • Mutagenic in Salmonella and mouse lymphoma assays • Available carcinogenicity studies in rats and hamsters were negative but these studies were inadequate (insufficient number of doses, and animals in rat and hamster studies; only one sex used in hamster study) • Higher priority for carcinogenicity testing should be assigned to allyl acetate; if allyl acetate proves to be carcinogenic, priority for carcinogenicity testing of allyl alcohol should be increased Priority: High • Date of Nomination: 4/1993 Table of Contents 1. Chemical Identification a. Basis of Nomination to the CSWG b. Input from Government Agencies/Industry c. Selection Status 2. Exposure Information 3. Evidence for Possible Carcinogenic Activity 4. References CHEMICAL IDENTIFICATION Allyl Acetate CAS Registry Number: 591-87-7 Chem. Abstracts Name: Acetic acid, 2-propenyl ester (9CI); Acetic acid, allyl ester (8CI) Synonyms and Trade Names: Allyl alcohol, acetate; 3-acetoxy-1-propene; 3-acetoxy- propene Allyl Alcohol CAS Registry Name: 107-18-6 Chemical Abstracts Name: 2-Propen-1-o1 (9CI) Synonyms and Trade Names: Allylic alcohol; 3-hydroxypropene; 1-propenol-3; 2-propenol; 2-propenyl alcohol; Shell unkrautted A; Weed Drench; vinyl carbinol Structure, Molecular Formula and Molecular Weight: Allyl Acetate C5H8O2 Mol. -

The Role of Oxides in Oxidation of Allyl Alcohol and Its Esters
CHEMISTRY & CHEMICAL TECHNOLOGY Chem. Chem. Technol., 2019, Chemistry Vol. 13, No. 1, pp. 46–51 THE ROLE OF OXIDES IN OXIDATION OF ALLYL ALCOHOL AND ITS ESTERS Eugeniy Fedevych1, Oleh Datsko2, Oleh Fedevych3, * https://doi.org/10.23939/chcht13.01.046 Abstract.1The assumption that oxides (glycidol, glycidol viscosity of biodiesel. It is a component of propellant [8] acetate, glycidol formate) are the intermediate molecular and possible power source during space flights [9]. products of the oxidation reaction of allyl alcohol and its Triacetin is used for the production of food plastics as esters in the medium of acetic acid has been grounded. To well as acrylic fabric filters for cigarettes due to its ability optimize the process of obtaining glycerin esters the to absorb carcinogenic substances. Since triacetin is kinetics of these oxides reaction with acetic and formic fermentation-resistant, it extends the shelf life of the acids has been investigated. Glycidol formate was found products and may be a component of confectioneries, to be the most stable compound and glycidol – the least including chocolate. Glycerin mono- and diacetin have stable one. The rate of oxides reaction with formic acid is similar properties [8]. by order higher than that with acetic acid. It is reasonable Taking into account the intense interest to the to use allyl formate as the starting material to achieve the above-mentioned compounds, we studied previously the maximum yields of glycerin esters. oxidative acylation of allyl acetate (AAc), allyl alcohol (AAl) and allyl formate (AF) and determined the Keywords: glycerin, allyl acetate, allyl formate, allyl composition of the resulting products [11]. -

(Cyclohexyloxy)Acetate, CAS Registry Number 68901-15-5 A.M
Food and Chemical Toxicology 82 (2015) S59–S65 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox RIFM fragrance ingredient safety assessment, allyl (cyclohexyloxy)acetate, CAS registry number 68901-15-5 A.M. Api a, D. Belsito b, S. Bhatia a, M. Bruze c, P. Calow d, M.L. Dagli e, W. Dekant f, A.D. Fryer g, L. Kromidas a,*, S. La Cava a, J.F. Lalko a, A. Lapczynski a, D.C. Liebler h, Y. Miyachi i, V.T. Politano a, G. Ritacco a, D. Salvito a, J. Shen a, T.W. Schultz j, I.G. Sipes k, B. Wall a, D.K. Wilcox a a Research Institute for Fragrance Materials, Inc., 50 Tice Boulevard, Woodcliff Lake, NJ 07677, USA b Member RIFM Expert Panel, Columbia University Medical Center, Department of Dermatology, 161 Fort Washington Ave., New York, NY 10032, USA c Member RIFM Expert Panel, Malmo University Hospital, Department of Occupational & Environmental Dermatology, Sodra Forstadsgatan 101, Entrance 47, Malmo SE-20502, Sweden d Member RIFM Expert Panel, University of Nebraska Lincoln, 230 Whittier Research Center, Lincoln, NE 68583-0857, USA e Member RIFM Expert Panel, University of Sao Paulo, School of Veterinary Medicine and Animal Science, Department of Pathology, Av. Prof. dr. Orlando Marques de Paiva, 87, Sao Paulo CEP 05508-900, Brazil f Member RIFM Expert Panel, University of Wuerzburg, Department of Toxicology, Versbacher Str. 9, 97078 Würzburg, Germany g Member RIFM Expert Panel, Oregon Health Science University, 3181 SW Sam Jackson Park Rd., Portland, OR 97239, USA h -
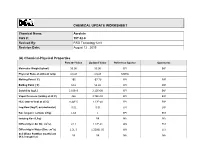
Acrolein CAS #: 107-02-8 Revised By: RRD Toxicology Unit Revision Date: August 12 , 2015
CHEMICAL UPDATE WORKSHEET Chemical Name: Acrolein CAS #: 107-02-8 Revised By: RRD Toxicology Unit Revision Date: August 12 , 2015 (A) Chemical-Physical Properties Part 201 Value Updated Value Reference Source Comments Molecular Weight (g/mol) 56.06 56.06 EPI EXP Physical State at ambient temp Liquid Liquid MDEQ Melting Point (˚C) 185 -87.70 EPI EXP Boiling Point (˚C) 52.6 52.60 EPI EXP Solubility (ug/L) 2.10E+8 2.12E+08 EPI EXP Vapor Pressure (mmHg at 25˚C) 266 2.74E+02 EPI EXP HLC (atm-m³/mol at 25˚C) 9.40E-5 1.22E-04 EPI EXP Log Kow (log P; octanol-water) -0.01 -0.01 EPI EXP Koc (organic carbon; L/Kg) 1.18 1 EPI EST Ionizing Koc (L/kg) NR NA NA Diffusivity in Air (Di; cm2/s) 0.11 1.12E-01 W9 EST Diffusivity in Water (Dw; cm2/s) 1.2E-5 1.2208E-05 W9 EST Soil Water Partition Coefficient NR NR NA NA (Kd; inorganics) CHEMICAL UPDATE WORKSHEET Acrolein (107-02-8) Part 201 Value Updated Value Reference Source Comments Flash Point (˚C) -15 F -26 CRC EXP Lower Explosivity Level (LEL; 0.028 0.028 CRC EXP unit less) Critical Temperature (K) 506.00 EPA2004 EXP Enthalpy of Vaporization 6.73E+03 EPA2004 EXP (cal/mol) Density (g/mL, g/cm3) 0.84 CRC EXP EMSOFT Flux Residential 2 m 2.43E-05 2.66E-05 EMSOFT EST (mg/day/cm2) EMSOFT Flux Residential 5 m 4.96E-05 5.98E-05 EMSOFT EST (mg/day/cm2) EMSOFT Flux Nonresidential 2 m 3.38E-05 4.18E-05 EMSOFT EST (mg/day/cm2) EMSOFT Flux Nonresidential 5 m 6.63E-05 9.12E-05 EMSOFT EST (mg/day/cm2) 2 CHEMICAL UPDATE WORKSHEET Acrolein (107-02-8) (B) Toxicity Values/Benchmarks Source/Reference/ Comments/Notes Part 201 Value Updated Value Date /Issues Reference Dose 1.6E-2 4.0E-3 ATSDR, 2007 (RfD) (mg/kg/day) Tier 2 Source: Basis: ATSDR intermediate oral MRL = 0.004 or 4.0E-3 mg/kg/day. -

Glycerol Ether Synthesis: a Bench Test for Green Chemistry Concepts
Glycerol Ether Synthesis: A Bench Test for Green Chemistry Concepts and Technologies Marc Sutter, Eric da Silva, Nicolas Duguet, Yann Raoul, Estelle Metay, Marc Lemaire To cite this version: Marc Sutter, Eric da Silva, Nicolas Duguet, Yann Raoul, Estelle Metay, et al.. Glycerol Ether Syn- thesis: A Bench Test for Green Chemistry Concepts and Technologies. Chemical Reviews, American Chemical Society, 2015, 115 (16), pp.8609-8651. 10.1021/cr5004002. hal-01312971 HAL Id: hal-01312971 https://hal.archives-ouvertes.fr/hal-01312971 Submitted on 3 Jun 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Glycerol Ether Synthesis: A Bench Test for Green Chemistry Concepts and Technologies Marc Sutter,a Eric Da Silva,a Nicolas Duguet,a Yann Raoul,b Estelle Métay,a Marc Lemairea,* a Equipe Catalyse Synthèse Environnement, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires, UMR-CNRS 5246, Université de Lyon, Université Claude Bernard-Lyon 1, Bâtiment Curien, 43 boulevard du 11 Novembre 1918, F-69622 Villeurbanne Cedex, France b Organisation Nationale Interprofessionnelle des Oléagineux, 11 rue de Monceau, CS 60003, 75378 PARIS CEDEX 08 Content 1. Introduction: properties and potential applications of glycerol ethers 2.