RECARBRIO, INN-Imipenem/Cilastatin/Relebactam
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

IDSA Guideline Review
1 Infectious Diseases Society of America Antimicrobial Resistant Treatment Guidance: Gram- Negative Bacterial Infections A Focus on Extended-Spectrum β-lactamase Producing Enterobacterales (ESBL-E), Carbapenem- Resistant Enterobacterales (CRE), and Pseudomonas aeruginosa with Difficult-to-Treat Resistance (DTR- P. aeruginosa) Guideline Summary by Kendra Suder, PharmD Candidate General Recommendations Considerations for choosing empiric therapy: o Local susceptibility patterns/ antibiogram o Patient’s previous organisms isolated & susceptibility data in the last 6 months o Antibiotic exposure in the last 30 days What if a patient was empirically placed on an antibiotic regimen that is now considered inactive against the organism based on susceptibility data? o If an inactive agent was started empirically for cystitis and patient improves, no change in antibiotic or extension of therapy is necessary o However, for other types of infections, guidelines recommend a change to an active regimen for a full treatment course (dated from the start of active therapy) Extended-Spectrum β-Lactamase-Producing Enterobacterales (ESBL-E) Extended-spectrum β-lactamase o Enzymes that inactivate most penicillins, cephalosporins, and aztreonam o Most commonly produced by Escherichia coli, Klebsiella pneumoniae, Klebsiella oxytoca, and Proteus mirabilis (guideline recommendations refer to ESBL-E infections caused by these organisms) o Most common type in the US is the CTX-M, especially CTX-M-15 In institutions that do not perform ESBL testing, a lack of -

Recarbrio, INN-Imipenem / Cilastatin / Relebactam
EMA/572792/2020 EMEA/H/C/004808 Recarbrio (imipenem / cilastatin / relebactam) An overview of Recarbrio and why it is authorised in the EU What is Recarbrio and what is it used for? Recarbrio is an antibiotic for treating adults with the following infections: • lung infections caught in hospital (hospital-acquired pneumonia), including ventilator-associated pneumonia (pneumonia caught while on a ventilator, which is a machine that helps a patient to breathe); • infection that has spread into the blood (bacteraemia) as a likely complication of hospital-acquired pneumonia or ventilator-associated pneumonia; • infections caused by bacteria classed as aerobic Gram-negative bacteria when other treatments might not work. Official guidance on the appropriate use of antibiotics should be considered when using the medicine. Recarbrio contains the active substances imipenem, cilastatin and relebactam. How is Recarbrio used? Recarbrio can only be obtained with a prescription and it should be used only after consulting a doctor with experience of managing infectious diseases. Recarbrio is given by infusion (drip) into a vein over 30 minutes. It is given every 6 hours for 5 to 14 days, depending on the nature of the infection. For more information about using Recarbrio, see the package leaflet or contact your doctor or pharmacist. How does Recarbrio work? One of the active substances in Recarbrio, imipenem, kills bacteria and the other two, cilastatin and relebactam, increase imipenem’s effectiveness in different ways. Imipenem interferes with bacterial proteins that are important for building the bacterial cell wall. This results in defective cell walls that collapse and cause the bacteria to die. -

New Β-Lactamase Inhibitor Combinations: Options for Treatment; Challenges for Testing
MEDICAL/SCIENTIFIC AffAIRS BULLETIN New β-lactamase Inhibitor Combinations: Options for Treatment; Challenges for Testing Background The β-lactam class of antimicrobial agents has played a crucial role in the treatment of infectious diseases since the discovery of penicillin, but β–lactamases (enzymes produced by the bacteria that can hydrolyze the β-lactam core of the antibiotic) have provided an ever expanding threat to their successful use. Over a thousand β-lactamases have been described. They can be divided into classes based on their molecular structure (Classes A, B, C and D) or their function (e.g., penicillinase, oxacillinase, extended-spectrum activity, or carbapenemase activity).1 While the first approach to addressing the problem ofβ -lactamases was to develop β-lactamase stable β-lactam antibiotics, such as extended-spectrum cephalosporins, another strategy that has emerged is to combine existing β-lactam antibiotics with β-lactamase inhibitors. Key β-lactam/β-lactamase inhibitor combinations that have been used widely for over a decade include amoxicillin/clavulanic acid, ampicillin/sulbactam, and pipercillin/tazobactam. The continued use of β-lactams has been threatened by the emergence and spread of extended-spectrum β-lactamases (ESBLs) and more recently by carbapenemases. The global spread of carbapenemase-producing organisms (CPOs) including Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baumannii, limits the use of all β-lactam agents, including extended-spectrum cephalosporins (e.g., cefotaxime, ceftriaxone, and ceftazidime) and the carbapenems (doripenem, ertapenem, imipenem, and meropenem). This has led to international concern and calls to action, including encouraging the development of new antimicrobial agents, enhancing infection prevention, and strengthening surveillance systems. -

AMEG Categorisation of Antibiotics
12 December 2019 EMA/CVMP/CHMP/682198/2017 Committee for Medicinal Products for Veterinary use (CVMP) Committee for Medicinal Products for Human Use (CHMP) Categorisation of antibiotics in the European Union Answer to the request from the European Commission for updating the scientific advice on the impact on public health and animal health of the use of antibiotics in animals Agreed by the Antimicrobial Advice ad hoc Expert Group (AMEG) 29 October 2018 Adopted by the CVMP for release for consultation 24 January 2019 Adopted by the CHMP for release for consultation 31 January 2019 Start of public consultation 5 February 2019 End of consultation (deadline for comments) 30 April 2019 Agreed by the Antimicrobial Advice ad hoc Expert Group (AMEG) 19 November 2019 Adopted by the CVMP 5 December 2019 Adopted by the CHMP 12 December 2019 Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2020. Reproduction is authorised provided the source is acknowledged. Categorisation of antibiotics in the European Union Table of Contents 1. Summary assessment and recommendations .......................................... 3 2. Introduction ............................................................................................ 7 2.1. Background ........................................................................................................ -

Title. 1 in Vitro Activity of the New Β-Lactamase Inhibitors Relebactam
bioRxiv preprint doi: https://doi.org/10.1101/499830; this version posted December 19, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Title. 2 In vitro activity of the new β-lactamase inhibitors relebactam and vaborbactam in combination 3 with β-lactams against Mycobacterium abscessus complex clinical isolates 4 5 Authors and affiliations. Amit Kaushik,a Nicole C. Ammerman,a Jin Lee,a Olumide Martins,a 6 Barry N. Kreiswirth,b Gyanu Lamichhane,a Nicole M. Parrish,c Eric L. Nuermbergera 7 8 aCenter for Tuberculosis Research, Johns Hopkins University School of Medicine, Baltimore, 9 Maryland, USA 10 bPublic Health Research Institute Tuberculosis Center, New Jersey Medical School - Rutgers, 11 The State University of New Jersey, Newark, New Jersey, USA 12 cDepartment of Pathology, Johns Hopkins University School of Medicine, Baltimore, Maryland, 13 USA 14 15 Key words. β-lactamase inhibitors, β-lactams, relebactam, vaborbactam, carbapenems, 16 cephalosporins, Mycobacterium abscessus 17 18 Running title. Relebactam/vaborbactam with β-lactams vs. M. abscessus Page 1 bioRxiv preprint doi: https://doi.org/10.1101/499830; this version posted December 19, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 19 Abstract 20 Pulmonary disease due to infection with Mycobacterium abscessus complex (MABC) is 21 notoriously difficult to treat, in large part due to MABC’s intrinsic resistance to most antibiotics, 22 including β-lactams. -

Antibiotics Currently in Clinical Development
A data table from Sept 2014 Antibiotics Currently in Clinical Development As of September 2014, an estimated 38 new antibiotics1 with the potential to treat serious bacterial infections are in clinical development for the U.S. market. The success rate for drug development is low; at best, only 1 in 5 candidates that enter human testing will be approved for patients.* This snapshot of the antibiotic pipeline will be updated periodically as products advance or are known to drop out of development. Please contact Rachel Zetts at [email protected] or 202-540-6557 with additions or updates. Cited for potential Development Known QIDP4 Drug name Company Drug class activity against gram- Potential indication(s)?5 phase2 designation? negative pathogens?3 Approved (for acute Acute bacterial skin and skin structure bacterial skin and skin infections, hospital-acquired bacterial Tedizolid Cubist Pharmaceuticals Oxazolidinone Yes structure infections), pneumonia/ventilator-acquired bacterial June 20, 2014 pneumonia Approved, May 23, Acute bacterial skin and skin structure Dalbavancin Durata Therapeutics Lipoglycopeptide Yes 2014 infections Approved, August 6, Acute bacterial skin and skin structure Oritavancin The Medicines Company Glycopeptide Yes 2014 infections New Drug Application (NDA) submitted (for Complicated urinary tract infections, complicated urinary complicated intra-abdominal infections, Novel cephalosporin+beta- Ceftolozane+tazobactam8 tract infection and Cubist Pharmaceuticals Yes Yes acute pyelonephritis (kidney infection), lactamase -

CLSI AST News Update Janet A
Volume 3, Issue 2 Spring 2018 CLSI Subcommittee on Antimicrobial Susceptibility Testing CLSI AST News Update Janet A. Hindler, MCLS MT(ASCP) F(AAM), Editor Audrey N. Schuetz, MD, MPH, D(ABMM), Editor The CLSI Outreach Working Group (ORWG) is providing this Newsletter to highlight some recent issues related to antimicrobial susceptibility testing Inside This Issue: and reporting. We are listing links to some new educational materials and reminding you where you can find information about the CLSI AST Featured Article: Part 1 Subcommittee proceedings. New β-lactam combination agents for the treatment of Gram-negative bacterial infections: what the clinical microbiologist needs to know! ..................................................4 Upcoming Webinar: Featured Article: Part 2 Why all the fuss over quality control of Preparation, Presentation, and Promotion of Cumulative Antibiograms To β-lactam combination agents? ........................8 Support Antimicrobial Stewardship Programs Case Study: Cefazolin, Urine, and October 16, 2018 | 1:00–2:00 PM Eastern (US) Time Escherichia coli, Klebsiella pneumoniae, and Presenters: Proteus mirabilis: Entertaining Solutions Sharon Erdman, PharmD for Antimicrobial Susceptibility Testing and Clinical Professor, Purdue University College of Pharmacy Reporting ..........................................................12 Infectious Diseases Clinical Pharmacist/Co-Director OPAT Program, Eskenazi Health Burning Question: What Should Clinical Laboratorians Know About Gonorrhea in Patricia J. Simner, PhD, D(ABMM) 2018? ................................................................15 Associate Professor of Pathology, Johns Hopkins University Director of Medical Bacteriology and Parasitology Laboratories, Johns Hopkins Hot Topic: It’s Enough to mec You Crazy! ...18 Hospital What does the CLSI AST Subcommittee do? The first edition of the CLSI AST News Update (Vol 1, Issue 1, Spring 2016) described details about the organization and operation of the CLSI AST Subcommittee. -
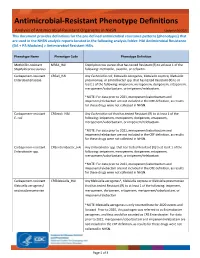
Antimicrobial Resistant Phenotype Definitions
Antimicrobial-Resistant Phenotype Definitions Analysis of Antimicrobial-Resistant Organisms in NHSN Updated 06/2021 This document provides definitions for the pre-defined antimicrobial resistance patterns (phenotypes) that are used in the NHSN analytic reports located in the following analysis folder: HAI Antimicrobial Resistance (DA + PA Modules) > Antimicrobial Resistant HAIs. Phenotype Name Phenotype Code Phenotype Definition Methicillin-resistant MRSA_HAI Staphylococcus aureus that has tested Resistant (R) to at least 1 of the Staphylococcus aureus following: methicillin, oxacillin, or cefoxitin. Carbapenem-resistant CREall_HAI Any Escherichia coli, Klebsiella aerogenes, Klebsiella oxytoca, Klebsiella Enterobacteriaceae pneumoniae, or Enterobacter spp. that has tested Resistant (R) to at least 1 of the following: imipenem, meropenem, doripenem, ertapenem, meropenem/vaborbactam, or imipenem/relebactam. *NOTE: For data prior to 2021, meropenem/vaborbactam and imipenem/relebactam are not included in the CRE definition, as results for these drugs were not collected in NHSN. Carbapenem-resistant CREecoli_HAI Any Escherichia coli that has tested Resistant (R) to at least 1 of the E. coli following: imipenem, meropenem, doripenem, ertapenem, meropenem/vaborbactam, or imipenem/relebactam. *NOTE: For data prior to 2021, meropenem/vaborbactam and imipenem/relebactam are not included in the CRE definition, as results for these drugs were not collected in NHSN. Carbapenem-resistant CREenterobacter_HAI Any Enterobacter spp. that has tested Resistant -

In Vitro Studies Evaluating the Activity of Imipenem in Combination with Relebactam Against Pseudomonas Aeruginosa Katherine Young1* , Ronald E
Young et al. BMC Microbiology (2019) 19:150 https://doi.org/10.1186/s12866-019-1522-7 RESEARCH ARTICLE Open Access In vitro studies evaluating the activity of imipenem in combination with relebactam against Pseudomonas aeruginosa Katherine Young1* , Ronald E. Painter1, Susan L. Raghoobar1, Nichelle N. Hairston1, Fred Racine1, Douglas Wisniewski1, Carl J. Balibar1, Artjohn Villafania1, Rumin Zhang1, Daniel F. Sahm2, Timothy Blizzard1, Nicholas Murgolo1, Milton L. Hammond1ˆ and Mary R. Motyl1 Abstract Background: The prevalence of antibiotic resistance is increasing, and multidrug-resistant Pseudomonas aeruginosa has been identified as a serious threat to human health. The production of β-lactamase is a key mechanism contributing to imipenem resistance in P. aeruginosa. Relebactam is a novel β-lactamase inhibitor, active against class A and C β-lactamases, that has been shown to restore imipenem susceptibility. In a series of studies, we assessed the interaction of relebactam with key mechanisms involved in carbapenem resistance in P. aeruginosa and to what extent relebactam might overcome imipenem non-susceptibility. Results: Relebactam demonstrated no intrinsic antibacterial activity against P. aeruginosa, had no inoculum effect, and was not subject to efflux. Enzymology studies showed relebactam is a potent (overall inhibition constant: 27 nM), practically irreversible inhibitor of P. aeruginosa AmpC. Among P. aeruginosa clinical isolates from the SMART global surveillance program (2009, n = 993; 2011, n = 1702; 2015, n = 5953; 2016, n = 6165), imipenem susceptibility rates were 68.4% in 2009, 67.4% in 2011, 70.4% in 2015, and 67.3% in 2016. With the addition of 4 μg/mL relebactam, imipenem susceptibility rates increased to 87.6, 86.0, 91.7, and 89.8%, respectively. -

Antibiotic Update
3/5/2015 Disclosure Antibiotic Update • I do not have a vested interest in or affiliation with any corporate organization offering financial support or grant monies for this Ashley Gustafson, Pharm.D., BCPS continuing education activity, or any affiliation PGY-2 Critical Care Pharmacy Resident with an organization whose philosophy could potentially bias my presentation Baptist Hospital of Miami Pharmacist Objectives Technician Objectives • Review antimicrobial resistance patterns • Recognize new antimicrobial agents and their appropriate use in infectious disease • Assess new antimicrobial agents and their appropriate indications • Discuss what is new in the antimicrobial pipeline • Discuss what is new in the antimicrobial pipeline • Explain why antimicrobial stewardship is • Explain why antimicrobial stewardship is important important Antibiotic Resistance Threats Antibiotic Resistance Variable Microbes Humans Factor “The microbes are educated to resist penicillin ...… In such No. on earth 5 x 10 31 6 x 10 9 ~ 10 22 cases the thoughtless person playing with penicillin is morally responsible for the death of the man who finally succumbs to Mass, metric ton 5 x 10 16 3 x 10 8 ~ 10 8 infection with the penicillin-resistant organism. I hope this evil 5 can be averted.” Generation time 30 min 30 years ~ 5 x 10 - Sir Alexander Fleming 1945 Time on earth, 3.5 x 10 9 4 x 10 6 ~ 10 3 years Spellberg B et al. CID 2008;46:155-64 1 3/5/2015 Antibiotic Resistance Antibiotic Pressure • Existed before antibiotics • Caused by 4 general mechanisms – -

Antimicrobial Use and Resistance (AUR) Module
February 2021 Antimicrobial Use and Resistance (AUR) Module Contents Antimicrobial Use and Resistance (AUR) Module ................................................................................. 1 Introduction .................................................................................................................................................. 1 1. Antimicrobial Use (AU) Option ................................................................................................................. 2 Introduction ......................................................................................................................................... 2 Requirements ....................................................................................................................................... 3 Data Analyses ....................................................................................................................................... 8 References .......................................................................................................................................... 14 Appendix A. Table of Instructions: Antimicrobial Use Option ........................................................... 15 Appendix B. List of Antimicrobials...................................................................................................... 17 Appendix C. Example Calculations of Antimicrobial Days .................................................................. 21 Appendix D: List of SAARsa ................................................................................................................ -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.