Analysis of Secreted Proteins from Adipocytes: a Proteomics Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evidence for an Active-Center Cysteine in the SH-Proteinase Cu-Clostripain Through Use of IV-Tosyl-L-Lysine Chloromethyl Ketone
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 173, number 1 FEBS 1649 July 1984 Evidence for an active-center cysteine in the SH-proteinase cu-clostripain through use of IV-tosyl-L-lysine chloromethyl ketone A.-M. Gilles and B. Keil Unitt! de Chimie des Protknes, Institut Pasteur, 28, rue du Docteur Roux, 75724 Paris CPdex 15, France Received 30 May 1984 The rapid reaction of a-clostripain with tosyl-L-lysine chloromethyl ketone results in a complete loss of activity and in the disappearance of one titratable SH group whereas the number of histidine residues is not affected. Tosyl-L-phenylalanine chloromethyl ketone and phenylmethylsulfonyl fluoride have no effect on the catalytic activity. From the molar ratio and under the assumption of 1: 1 molar interaction, the fully active enzyme has a specific activity of 650-700 units/mg [twice the value proposed by Porter et al. (J. Biol. Chem. 246 (1971) 76757682)]. Partial oxidation makes it experimentally impossible to attain this maximal value. ff-Clostripain Cysteine proteinase Active site 1. INTRODUCTION was due to the modification of a thiol group in an analogous way with other cysteine proteinases such Clostripain (EC 3.4.4.20) is a sulfhydryl protein- as papain [6] and ficin [7]. Recently [8], we eluci- ase isolated from the culture filtrate of Clostridium dated the amino acid sequence around this acces- histolyticum with a highly limited specificity sible thiol group after labelling with radioactive directed at the carboxyl bond of arginyl residues in iodoacetic acid. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

The Proteolysis of Apolipoprotein E in Alzheimer's Disease
THE PROTEOLYSIS OF APOLIPOPROTEIN E IN ALZHEIMER’S DISEASE by Julia Love A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Biology Boise State University August 2016 © 2016 Julia Love ALL RIGHTS RESERVED BOISE STATE UNIVERSITY GRADUATE COLLEGE DEFENSE COMMITTEE AND FINAL READING APPROVALS of the thesis submitted by Julia Love Thesis Title: The Proteolysis of Apolipoprotein E in Alzheimer’s Disease Date of Final Oral Examination: 26 April 2016 The following individuals read and discussed the thesis submitted by student Julia Love, and they evaluated her presentation and response to questions during the final oral examination. They found that the student passed the final oral examination. Troy Rohn, Ph.D. Chair, Supervisory Committee Kenneth A. Cornell, Ph.D. Member, Supervisory Committee Juliette Tinker, Ph.D. Member, Supervisory Committee The final reading approval of the thesis was granted by Troy Rohn, Ph.D., Chair of the Supervisory Committee. The thesis was approved for the Graduate College by Jodi Chilson, M.F.A., Coordinator of Theses and Dissertations. DEDICATION This thesis is dedicated to my parents Paul and Cynthia Love, my brother Philip Love, and all of my friends who have supported and encouraged me along the way. iv ACKNOWLEDGEMENTS There have been many people who have contributed to this work and my academic growth over the course of pursuing my Master’s degree. These individual contributions have not gone unnoticed and are an important part of my thesis work. First and foremost, I would like to thank Dr. Troy Rohn for being available with a willing attitude whenever I needed assistance, for his steadfast support and care, and for providing me with every opportunity to exceed what I thought were my limitations. -

Proteolytic Enzymes in Grass Pollen and Their Relationship to Allergenic Proteins
Proteolytic Enzymes in Grass Pollen and their Relationship to Allergenic Proteins By Rohit G. Saldanha A thesis submitted in fulfilment of the requirements for the degree of Masters by Research Faculty of Medicine The University of New South Wales March 2005 TABLE OF CONTENTS TABLE OF CONTENTS 1 LIST OF FIGURES 6 LIST OF TABLES 8 LIST OF TABLES 8 ABBREVIATIONS 8 ACKNOWLEDGEMENTS 11 PUBLISHED WORK FROM THIS THESIS 12 ABSTRACT 13 1. ASTHMA AND SENSITISATION IN ALLERGIC DISEASES 14 1.1 Defining Asthma and its Clinical Presentation 14 1.2 Inflammatory Responses in Asthma 15 1.2.1 The Early Phase Response 15 1.2.2 The Late Phase Reaction 16 1.3 Effects of Airway Inflammation 16 1.3.1 Respiratory Epithelium 16 1.3.2 Airway Remodelling 17 1.4 Classification of Asthma 18 1.4.1 Extrinsic Asthma 19 1.4.2 Intrinsic Asthma 19 1.5 Prevalence of Asthma 20 1.6 Immunological Sensitisation 22 1.7 Antigen Presentation and development of T cell Responses. 22 1.8 Factors Influencing T cell Activation Responses 25 1.8.1 Co-Stimulatory Interactions 25 1.8.2 Cognate Cellular Interactions 26 1.8.3 Soluble Pro-inflammatory Factors 26 1.9 Intracellular Signalling Mechanisms Regulating T cell Differentiation 30 2 POLLEN ALLERGENS AND THEIR RELATIONSHIP TO PROTEOLYTIC ENZYMES 33 1 2.1 The Role of Pollen Allergens in Asthma 33 2.2 Environmental Factors influencing Pollen Exposure 33 2.3 Classification of Pollen Sources 35 2.3.1 Taxonomy of Pollen Sources 35 2.3.2 Cross-Reactivity between different Pollen Allergens 40 2.4 Classification of Pollen Allergens 41 2.4.1 -

Durham E-Theses
Durham E-Theses Midgut proteases from larval spodoptera littoralis (lepidoptera: noctutoae) Lee, Michael James How to cite: Lee, Michael James (1992) Midgut proteases from larval spodoptera littoralis (lepidoptera: noctutoae), Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/5739/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk MIDGUT PROTEASES FROM LARVAL SPODOPTERA LITTORALIS (LEPIDOPTERA: NOCTUTOAE) By Michael James Lee B.Sc. (Dunelm) The copyright of this thesis rests with the author. No quotation from it should be pubhshed without his prior written consent and information derived from it should be acknowledged. Being a thesis submitted for the degree of Doctor of Philosophy of the University of Durham. November, 1992 Hatfield College University of Durham 6 APR 1993 DECLARATION I hereby declare that the work presented in this document is based on research carried out by me, and that no part has been previously submitted for a degree in this or any other university. -

Enzymes for Cell Dissociation and Lysis
Issue 2, 2006 FOR LIFE SCIENCE RESEARCH DETACHMENT OF CULTURED CELLS LYSIS AND PROTOPLAST PREPARATION OF: Yeast Bacteria Plant Cells PERMEABILIZATION OF MAMMALIAN CELLS MITOCHONDRIA ISOLATION Schematic representation of plant and bacterial cell wall structure. Foreground: Plant cell wall structure Background: Bacterial cell wall structure Enzymes for Cell Dissociation and Lysis sigma-aldrich.com The Sigma Aldrich Web site offers several new tools to help fuel your metabolomics and nutrition research FOR LIFE SCIENCE RESEARCH Issue 2, 2006 Sigma-Aldrich Corporation 3050 Spruce Avenue St. Louis, MO 63103 Table of Contents The new Metabolomics Resource Center at: Enzymes for Cell Dissociation and Lysis sigma-aldrich.com/metpath Sigma-Aldrich is proud of our continuing alliance with the Enzymes for Cell Detachment International Union of Biochemistry and Molecular Biology. Together and Tissue Dissociation Collagenase ..........................................................1 we produce, animate and publish the Nicholson Metabolic Pathway Hyaluronidase ...................................................... 7 Charts, created and continually updated by Dr. Donald Nicholson. DNase ................................................................. 8 These classic resources can be downloaded from the Sigma-Aldrich Elastase ............................................................... 9 Web site as PDF or GIF files at no charge. This site also features our Papain ................................................................10 Protease Type XIV -

Degradative Inactivation of Cyclic AMP-Dependent Protein
Proc. Natl Acad. Sci. USA Vol. 78, No. 6, pp. 3492-3495, June 1981 Biochemistry Degradative inactivation of cyclic AMP-dependent protein kinase by a membranal proteinase is restricted to the free catalytic subunit in its native conformation (brush-border membranes/intestinal microvilli/enzyme regulation/limited proteolysis) EYTAN ALHANATY, JONATHAN PATINKIN, MIRIAM TAUBER-FINKELSTEIN, AND SHMUEL SHALTIEL* Department of Chemical Immunology, The Weizmann Institute of Science, Rehovot, Israel Communicated by Michael Sela, March 13, 1981 ABSTRACT A membranal proteinase from brush-border ep- This raises the possibility that there may be additional regula- ithelial cells of the rat small intestine was shown to bring about a tory devices for modulating the cellular response to the hor- restricted and limited degradation ofthe free catalytic subunit (C) monal stimulus. of cyclic AMP-dependent protein kinase (ATP:protein phospho- cAMPdPKase activity in brush-border membranes (from the transferase, EC 2.7.1.37) with concomitant inactivation of the ki- rat small intestine) vanishes within a few minutes upon addition nase. This membranal proteinase exhibits a remarkable specific- of cAMP (10). The inactivation was shown to be due to the ex- ity. (i) It degrades C in its native conformation, but not after it has istence in these membranes of an enzyme that brings about a been heat-denatured. (ii) The degradation of C (Mr 40,000) does specific, limited degradation of the catalytic subunit of not proceed further, once a distinct clipped product (Mr 34,000) did not attack (under is formed. (iii) The undissociated ("stored") form of the enzyme cAMPdPKase. This membranal enzyme (R2C2) is not attacked by the membranal proteinase, preserving the same conditions) other proteins in the membrane prepa- both its potential catalytic activity and its molecular integrity. -

A Cysteine Proteinase Inhibitor of Human Saliva We Have Recently
COMMUNICATION J. Biochem. 96,1311-1314 (1984) Cystatin S : A Cysteine Proteinase Inhibitor of Human Saliva Satoko ISEMURA,* Eiichi SAITOH,* Seiki ITO,** Mamoru ISEMURA,*** and Kazuo SANADA* *Department of Oral Biochemistry , Niigata Faculty, Nippon Dental University. Niigata 951, **First Department of Internal Medicine, Niigata University School of Medicine, Niigata 951, and ***Department of Biochemistry, Tohoku University School of Medicine, Sendai, Miyagi 980 Received for publication, July 12, 1984 An acidic protein of human saliva, which we named SAP-1 previously, is now shown to be an inhibitor of several cysteine proteinases. The protein inhibited papain and ficin strongly, and stem bromelain and bovine cathepsin C partially. How ever, it did not inhibit either porcine cathepsin B or clostripain. The mode of the inhibition of papain was found to be non-competitive. The name cystatin S has been proposed for this salivary protein in view of the similarities in activity and structure to other cysteine proteinase inhibitors such as chicken egg-white cystatin and human cystatins A, B, and C. The cystatin S antigen was detected immunohistochemically in the serous cells of human parotid and submaxillary glands. We have recently isolated an acidic protein, SAP-1, 3.4.22.3], bromelain [EC 3.4.22.4], and clostripain with a molecular weight of 12,552 and PI 4.68 [EC 3.4.22.8] from Sigma; cathepsin C [dipeptidyl from human whole saliva, and determined its peptidase I, EC 3.4.14.1] from Serva; trypsin [EC amino acid sequence (1). This protein has se 3.4.21.4] and chymotrypsin [EC 3.4.21.1] from quence homology of 54% with human y-trace, the Worthington Biochemical Inc. -
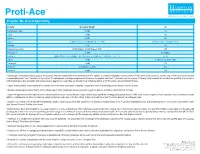
Proti-Ace Enzyme Mr Pi Specificty Source
Proti-Ace TM Solutions for Crystal Growth Enzyme, Mr, pI and Specificity Enzyme Molecular Weight pI Chymotrypsin, Alpha 25,000 9.1 Trypsin 23,800 10.5 Elastase 25,900 8.5 Papain 23,400 (Theoretical), 23,000 (Dreuth et al. 1968) 6.8(Theoretical), 8.75 (Experimental) Subtilisin 27,287 9.4 Endoproteinase Glu-C 29,020 (Sigma), 27,000 (Drapeau 1978) N/A Proteinase K 28,900 8.9 (Sigma) Clostripain 50,000 (Mitchell and Harrington 1968), Two chains of 45,000 and 12,500 (Gilles et al. 1978) 4.8-4.9 Pepsin 34,600 1.0 (Bovey and Yanari 1960) Thermolysin 34,600 4.45 Bromelain 22,500 (Wharton 1974) 9.55 Actinase E 20,000 N/A • Chymotrypsin preferentially cleaves peptide amide bonds where the carboxyl side of the amide bond (the P1 position) is a tyrosine, tryptophan, or phenylalanine. These amino acids contain an aromatic ring in their side chain that fits into a ‘hydrophobic pocket’ (the S1 position) of the enzyme. The hydrophobic and shape complementarity between the peptide substrate P1 side chain and the enzyme S1 binding cavity accounts for the substrate specificity of this enzyme. [2][3] Chymotrypsin also hydrolyzes other amide bonds in peptides at slower rates, particularly those containing leucine at the P1 position. Source: Bovine Pancreas • Trypsin cleaves peptide chains mainly at the carboxyl side of the amino acids lysine or arginine, except when either is followed by proline. Source: Bovine Pancreas • Elastase cleaves peptide chains mainly at the carboxy side of small, hydrophobic amino acids such as glycine, alanine, and valine. -

A New Insight Into Phagocytosis of Apoptotic Cells: Proteolytic Enzymes Divert the Recognition and Clearance of Polymorphonuclear Leukocytes by Macrophages
Cell Death and Differentiation (2007) 14, 171–182 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd A new insight into phagocytosis of apoptotic cells: proteolytic enzymes divert the recognition and clearance of polymorphonuclear leukocytes by macrophages K Guzik1, M Bzowska1, J Smagur2, O Krupa1,2, M Sieprawska2, J Travis3 and J Potempa*,2,3 The recognition of phosphatidylserine (PS) on the surface of any apoptotic cell is considered to be a key event for its clearance. We challenge this concept by showing that pretreatment of neutrophils with either host or bacterial protease affects their uptake by human monocyte-derived macrophages without having an effect on cell-surface PS presentation. Specifically, whereas preincubation of apoptotic neutrophils with cathepsin G or thrombin significantly inhibited their uptake, gingipains R or clostripain enhanced phagocytosis by macrophages. Moreover, bacterial proteinases sensitized healthy neutrophils for uptake by macrophages, whereas endogenous proteinases were unable to elicit this effect. This stimulation was apparently owing to the combined effect of proteolytic cleavage of an antiphagocytic signal (CD31) and the generation of a novel ‘eat-me’ signal on the neutrophil surface. These results argue that neutrophil recognition and phagocytosis by macrophages is mediated by a protein ligand whose proteolytic modification could affect the local inflammatory process. Cell Death and Differentiation (2007) 14, 171–182. doi:10.1038/sj.cdd.4401927; published online 21 April 2006 Successful uptake of an apoptotic cell requires recognition of TSP and collectins (e.g. mannose binding lectin, surfactant ‘eat-me’ signals exposed on the surface of the dying cell by protein A, and C1q).6 Together with the panel of bridging specific receptors on macrophages. -

Families and Clans of Cysteine Peptidases
Families and clans of eysteine peptidases Alan J. Barrett* and Neil D. Rawlings Peptidase Laboratory. Department of Immunology, The Babraham Institute, Cambridge CB2 4AT,, UK. Summary The known cysteine peptidases have been classified into 35 sequence families. We argue that these have arisen from at least five separate evolutionary origins, each of which is represented by a set of one or more modern-day families, termed a clan. Clan CA is the largest, containing the papain family, C1, and others with the Cys/His catalytic dyad. Clan CB (His/Cys dyad) contains enzymes from RNA viruses that are distantly related to chymotrypsin. The peptidases of clan CC are also from RNA viruses, but have papain-like Cys/His catalytic sites. Clans CD and CE contain only one family each, those of interleukin-ll3-converting enz3wne and adenovirus L3 proteinase, respectively. A few families cannot yet be assigned to clans. In view of the number of separate origins of enzymes of this type, one should be cautious in generalising about the catalytic mechanisms and other properties of cysteine peptidases as a whole. In contrast, it may be safer to gener- alise for enzymes within a single family or clan. Introduction Peptidases in which the thiol group of a cysteine residue serves as the nucleophile in catalysis are defined as cysteine peptidases. In all the cysteine peptidases discovered so far, the activity depends upon a catalytic dyad, the second member of which is a histidine residue acting as a general base. The majority of cysteine peptidases are endopeptidases, but some act additionally or exclusively as exopeptidases. -

CHAPTER V General Discussion
CHAPTER V General Discussion GENERAL DISCUSSION The cysteine endopeptidases represent one of the four classes of enzymes that act on peptide bonds of proteins and oligopeptides. Papain, the protogonist of the cysteine endopeptidases has been by far the most extensively studied of this class of enzymes. Most of the cysteine endopeptidases characterised so far show a high degree of similarity with regard to their physico-chemical properties, specificity and primary and secondary structures to papain, and are now recognised as papain superfamily. Rawlings and Barrett, (1993) have classified endopeptidases into different families based on the sequence homology and active site residues. They showed that there are 14 different families of cysteine endopeptidases and the papain family is the largest. In the absence of sequence data, kinetic parameters of inhibitor binding and knowledge of active site residues provides ample scope to classify endopeptidases at least into papain and nonpapain families. Hence, based on these information an attempt was made to classify the cysteine endopeptidases investigated in these studies. Vignain, legumain, glycylendopeptidase (papaya proteinase IV) and clostripain are activated in the presence of thiols and have a pH optimum of 5-7, a general characteristics for enzymes belonging to the cysteine class. The molecular weight of enzymes belonging to the papain family fall in the range of 23 kDa - 28 kDa with papain having a molecular weight of 23.35 kDa. Vignain (28 kDa) and glycylendopeptidase (25 kDa) have molecular weights in this range, whereas clostripain and legumain have a molecular weights of 58 kDa and 33 kDa, respectively, which are higher than that of papain.