Deciphering the Genome Repertoire of Pseudomonas Sp. M1 Toward B-Myrcene Biotransformation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drug Metabolism Chemical and Enzymatic Aspects Textbook Edition
DRUG METABOLISM Chemical and Enzymatic Aspects TEXTBOOK EDITION Jack P. Uetrecht University of Toronto Ontario, Canada William Trager University of Washington Seattle, Washington, USA Uetrecht_978-1420061031_TP.indd 2 5/11/07 2:28:26 PM OTE/SPH OTE/SPH uetrecht IHUS001-Uetrecht May 10, 2007 3:29 Char Count= Informa Healthcare USA, Inc. 52 Vanderbilt Avenue New York, NY 10017 C 2007 by Informa Healthcare USA, Inc. Informa Healthcare is an Informa business No claim to original U.S. Government works Printed in the United States of America on acid-free paper 10987654321 International Standard Book Number-10: 1-4200-6103-8 International Standard Book Number-13: 978-1-4200-6103-1 This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. A wide variety of references are listed. Reasonable efforts have been made to publish reliable data and information, but the author and the publisher cannot assume responsibility for the validity of all materials or for the consequence of their use. No part of this book may be reprinted, reproduced, transmitted, or utilized in any form by any electronic, mechan- ical, or other means, now known or hereafter invented, including photocopying, microfilming, and recording, or in any information storage or retrieval system, without written permission from the publishers For permission to photocopy or use material electronically from this work, please access www.copyright.com (http://www.copyright.com/) or contact the Copyright Clearance Center, Inc. (CCC) 222 Rosewood Drive, Danvers, MA 01923, 978-750-8400. CCC is a not-for-profit organization that provides licenses and registra- tion for a variety of users. -

Acyclic Monoterpene Primary Alcohol :NADP+ Oxidoreductase of Rauwolfia Serpentina Cells: the Key Enzyme in Biosynthesis of Monoterpene Alcohols
J. Biochem. 109, 341-347 (1991) Acyclic Monoterpene Primary Alcohol :NADP+ Oxidoreductase of Rauwolfia serpentina Cells: The Key Enzyme in Biosynthesis of Monoterpene Alcohols Hiromitsu Ikeda, * Nobuyoshi Esaki, ** Shunji Nakai, * Keij i Hashimoto, Shinichi Uesato,*•' Kenji Soda,** and Tetsuro Fujita*2 *Department of Natural Product Chemistry , Faculty of Pharmaceutical Sciences,Kyoto University,Sakyo-ku, Kyoto, Kyoto 606; **Laboratory of Microbial Biochemistry,Institute for ChemicalResearch, Kyoto University, Uji, Kyoto 611; and *`'Kyoto Pharmaceutical University, Yamashina-ku, Kyoto, Kyoto 607 Received for publication, August 1, 1990 Acyclic monoterpene primary alcohol: NADP+ oxidoreductase, a key enzyme in the biosynthesis of monoterpene alcohols in plants, is unstable and has been only poorly characterized. However we have established conditions which stabilize the enzyme from Rauwolfza serpentina cells, and then purified it to homogeneity. It is a monomer with a molecular weight of about 44,000 and contains zinc ions. Various branched-chain allylic primary alcohols such as nerol, geraniol, and 10-hydroxygeraniol were substrates, but ethanol was inert. The enzyme exclusively requires NADP+ or NADPH as the cofactor. Steady-state kinetic studies showed that the nerol dehydrogenation proceeds by an ordered Bi-Bi mechanism. NADP+ binds the enzyme first and then NADPH is the second product released from it. Gas chromatography-mass spectrometric analysis of the reaction products showed that 10-hydroxygeraniol undergoes a reversible dehydrogenation to produce 10-oxogeraniol or 10-hydroxygeranial, which are oxidized further to give 10-oxogeranial, the direct precursor of iridodial. The enzyme has been found to exclusively transfer the pro-R hydrogen of NADPH to neral. The N-terminal sequence of the first 21 amino acids revealed no significant homology with those of various other proteins including the NAD(P)+-dependent alcohol dehydrogenases registered in a protein data bank. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Physiology of Deletion Mutants in the Anaerobic Β-Myrcene Degradation Pathway in Castellaniella Defragrans Frauke Lüddeke, Aytac Dikfidan and Jens Harder*
Lüddeke et al. BMC Microbiology 2012, 12:192 http://www.biomedcentral.com/1471-2180/12/192 RESEARCH ARTICLE Open Access Physiology of deletion mutants in the anaerobic β-myrcene degradation pathway in Castellaniella defragrans Frauke Lüddeke, Aytac Dikfidan and Jens Harder* Abstract Background: Monoterpenes present a large and versatile group of unsaturated hydrocarbons of plant origin with widespread use in the fragrance as well as food industry. The anaerobic β-myrcene degradation pathway in Castellaniella defragrans strain 65Phen differs from well known aerobic, monooxygenase-containing pathways. The initial enzyme linalool dehydratase-isomerase ldi/LDI catalyzes the hydration of β-myrcene to (S)-(+)-linalool and its isomerization to geraniol. A high-affinity geraniol dehydrogenase geoA/GeDH and a geranial dehydrogenase geoB/ GaDH contribute to the formation of geranic acid. A genetic system was for the first time applied for the betaproteobacterium to prove in vivo the relevance of the linalool dehydratase-isomerase and the geraniol dehydrogenase. In-frame deletion cassettes were introduced by conjugation and two homologous recombination events. Results: Polar effects were absent in the in-frame deletion mutants C. defragrans Δldi and C. defragrans ΔgeoA. The physiological characterization of the strains demonstrated a requirement of the linalool dehydratase-isomerase for growth on acyclic monoterpenes, but not on cyclic monoterpenes. The deletion of geoA resulted in a phenotype with hampered growth rate on monoterpenes as sole carbon and energy source as well as reduced biomass yields. Enzyme assays revealed the presence of a second geraniol dehydrogenase. The deletion mutants were in trans complemented with the broad-host range expression vector pBBR1MCS-4ldi and pBBR1MCS-2geoA, restoring in both cases the wild type phenotype. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -
Geraniol and Geranial Dehydrogenases Induced in Anaerobic Monoterpene Degradation by Castellaniella Defragrans
Geraniol and Geranial Dehydrogenases Induced in Anaerobic Monoterpene Degradation by Castellaniella defragrans Frauke Lüddeke,a Annika Wülfing,a Markus Timke,a Frauke Germer,a Johanna Weber,a Aytac Dikfidan,a Tobias Rahnfeld,a Dietmar Linder,c Anke Meyerdierks,b and Jens Hardera Department of Microbiologya and Department of Molecular Ecology,b Max Planck Institute for Marine Microbiology, Bremen, Germany, and Biochemisches Institut, Fachbereich Medizin, Justus-Liebig-Universität, Giessen, Germanyc Castellaniella defragrans is a Betaproteobacterium capable of coupling the oxidation of monoterpenes with denitrification. Ge- raniol dehydrogenase (GeDH) activity was induced during growth with limonene in comparison to growth with acetate. The Downloaded from N-terminal sequence of the purified enzyme directed the cloning of the corresponding open reading frame (ORF), the first bacte- rial gene for a GeDH (geoA, for geraniol oxidation pathway). The C. defragrans geraniol dehydrogenase is a homodimeric en- zyme that affiliates with the zinc-containing benzyl alcohol dehydrogenases in the superfamily of medium-chain-length dehy- ؋ ؍ drogenases/reductases (MDR). The purified enzyme most efficiently catalyzes the oxidation of perillyl alcohol (kcat/Km 2.02 ؋ 6 ؊1 ؊1 ؍ ؊1 ؊1 6 10 M s ), followed by geraniol (kcat/Km 1.57 10 M s ). Apparent Km values of <10 M are consistent with an in vivo toxicity of geraniol above 5 M. In the genetic vicinity of geoA is a putative aldehyde dehydrogenase that was named geoB and identified as a highly abundant protein during growth with phellandrene. Extracts of Escherichia coli expressing geoB demon- strated in vitro a geranial dehydrogenase (GaDH) activity. GaDH activity was independent of coenzyme A. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -

Sterol Biosynthesis Pathway Is Part of the Interferon Host Defence Response
Sterol biosynthesis pathway is part of the interferon host defence response Mathieu Blanc A thesis submitted in fulfilment of requirements for the degree of Doctor of Philosophy Division of Pathway Medicine, School of Biomedical Sciences The University of Edinburgh April 2010 Declaration I hereby declare that this thesis is of my own composition, and that it contains no material previously submitted for the award of any other degree. The work reported in this thesis has been executed by myself, except where due acknowledgement is made in the text. Mathieu Blanc ii Abstract Recently, cholesterol metabolism has been shown to modulate the infection of several viruses and there is growing evidence that inflammatory response to infection also modulates lipid metabolism. However little is known about the role of inflammatory processes in modulating lipid metabolism and their consequences for the viral infection. This study investigates host-lipid viral interaction pathways using mouse cytomegalovirus, a large double-stranded DNA genome, which represents one of the few models for a natural infection of its natural host. In this study, transcriptomic and lipidomic profiling of macrophages shows that there is a specific coordinated regulation of the sterol pathways upon viral infection or treatment with IFN or (but not TNFα, IL1 or IL6) resulting in the decrease of free cellular cholesterol. Furthermore, we show that pharmacological and RNAi inhibition of the sterol pathway augments protection against infection in vitro and in vivo and we identified that the prenylation branch of the sterol metabolic network was involved in the protective response. Finally, we show that genetic knock out of IFN results in a partial reduction while genetic knock out of Ifnar1 completely abolishes the reduction of the sterol biosynthetic activity upon infection. -

Differential Expression and Functional Analysis of Two Short-Chain Alcohol
FOLIA HORTICULTURAE Folia Hort. 32(1) (2020): 97–114 Published by the Polish Society DOI: 10.2478/fhort-2020-0010 for Horticultural Science since 1989 RESEARCH ARTICLE Open access http://www.foliahort.ogr.ur.krakow.pl Differential expression and functional analysis of two short-chain alcohol dehydrogenases/reductases in Hedychium coronarium Hua Chen1, Yuechong Yue2,3, Rangcai Yu2,3, Yanping Fan2,3,* 1 Department of Landscape Architecture, College of Life Science, Zhaoqing University, Zhaoqing 526061, China 2 The Research Center for Ornamental Plants, College of Forestry and Landscape Architecture, South China Agricultural University, Guangzhou 510642, China 3 Guangdong Key Laboratory for Innovative Development and Utilization of Forest Plant Germplasm, South China Agricultural University, Guangzhou 510642, China ABSTRACT In this study, the full cDNA sequences of HcADH2 and HcADH3 were cloned from Hedychium coronarium. The amino acid sequences encoded by them contained three most conserved motifs of short-chain alcohol dehydrogenase (ADH), namely NAD+ binding domain, TGxxx[AG]xG and active site YxxxK. The highest similarity between two genes and ADH from other plants was 70%. Phylogenetic analysis showed that they belonged to a member of the short-chain dehydrogenases/ reductases 110C subfamily, but they were distinctly clustered in different clades. Real-time polymerase chain reaction analyses showed that HcADH2 was specifically expressed in bract, and it was expressed higher in no-scented Hedychium forrestii than other Hedychium species, but was undetectable in Hedychium coccineum. HcADH3 was expressed higher in the lateral petal of the flower than in other vegetative organs, and it was expressed the most in H. coronarium that is the most scented among Hedychium species, and its expression levels peaked at the half opening stage. -

Title Two Types of Alcohol Dehydrogenase from Perilla Can Form Citral and Perillaldehyde
Two types of alcohol dehydrogenase from Perilla can form Title citral and perillaldehyde. Author(s) Sato-Masumoto, Naoko; Ito, Michiho Citation Phytochemistry (2014), 104: 12-20 Issue Date 2014-08 URL http://hdl.handle.net/2433/187787 Right © 2014 Elsevier Ltd. Type Journal Article Textversion author Kyoto University Title Two types of alcohol dehydrogenase from Perilla can form citral and perillaldehyde Naoko Sato-Masumoto and Michiho Ito* Abstract Studies on the biosynthesis of oil compounds in Perilla will help in understanding regulatory systems of secondary metabolites and in elucidating reaction mechanisms for natural product synthesis. In this study, two types of alcohol dehydrogenases, an aldo-keto reductase (AKR) and a geraniol dehydrogenase (GeDH), which are thought to participate in the biosynthesis of perilla essential oil components such as citral and perillaldehyde, were isolated from three pure lines of perilla. These enzymes shared high amino acid sequence identity within genus Perilla, and were expressed regardless of oil type. The overall reaction from geranyl diphosphate to citral was performed in vitro using geraniol synthase and GeDH to form a large proportion of citral and relatively little geraniol as reaction products. The biosynthetic pathway from geranyl diphosphate to citral, the main compound of citral-type perilla essential oil, was revealed in this study. Keywords Perilla; Labiatae; aldo-keto reductase; geraniol dehydrogenase; geraniol; citral; sequential enzymatic reaction; biosynthetic pathway; Essential oil 1 1. Introduction Japanese perilla plants are classified into four species including a cultivated species, Perilla frutescens, and three wild species, P. citriodora, P. hirtella, and P. setoyensis (Ito and Honda, 1996). Plants of each species contain essential oil that is composed principally of unique monoterpene or phenylpropene compounds. -

(19) United States (12) Patent Application Publication (10) Pub
US 20130244920A1 (19) United States (12) Patent Application Publication (10) Pub. N0.: US 2013/0244920 A1 Lee et al. (43) Pub. Date: Sep. 19, 2013 (54) WATER SOLUBLE COMPOSITIONS (52) US. Cl. INCORPORATING ENZYMES, AND METHOD USPC ......................................... .. 510/392; 264/299 OF MAKING SAME (57) ABSTRACT (76) Inventors: David M. Lee, CroWn Point, IN (US); Jennifer L‘ Sims’ Lowell’ IN (Us) Disclosed herein are Water soluble compositions, such as ?lms, including a mixture of a ?rst Water-soluble resin, an (21) Appl' NO': 13/422’709 enzyme, and an enzyme stabilizer Which comprises a func (22) Filed: Man 16, 2012 tional substrate for the enzyme, methods of making such compositions, and methods of using such compositions, e.g. Publication Classi?cation to make packets containing functional ingredients. The enzymes can include proteases and mixtures of proteases (51) Int. Cl. With other enzymes, and the compositions provide good C11D 3/386 (2006.01) retention of enzyme function following ?lm processing and B29C 39/02 (2006.01) storage. US 2013/0244920 A1 Sep. 19,2013 WATER SOLUBLE COMPOSITIONS preheated to a temperature less than 77° C., optionally in a INCORPORATING ENZYMES, AND METHOD range ofabout 66° C. to about 77° C., or about 74° C.; drying OF MAKING SAME the Water from the cast mixture over a period of less than 24 hours, optionally less than 12 hours, optionally less than 8 FIELD OF THE DISCLOSURE hours, optionally less than 2 hours, optionally less than 1 [0001] The present disclosure relates generally to Water hour, optionally less than 45 minutes, optionally less than 30 soluble ?lms. -
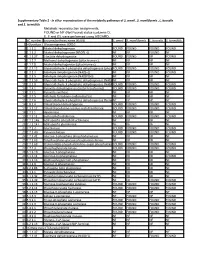
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase.