Cyproterone Acetate Acts As a Disruptor of the Aryl Hydrocarbon Receptor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
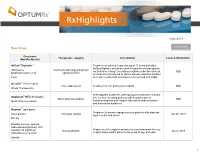
July 2019 New Drugs
July 2019 Learn more New drugs Drug name Therapeutic category Indication(s) Launch information Manufacturer(s) AirDuo® Digihaler™ Treatment of asthma in patients aged 12 years and older. AirDuo Digihaler should be used for patients not adequately (fluticasone Corticosteroid/ long-acting beta controlled on a long term asthma control medication such as TBD propionate/salmeterol) agonist (LABA) an inhaled corticosteroid or whose disease warrants initiation Teva of treatment with both an inhaled corticosteroid and LABA Accrufer® (ferric maltol) Iron replacement Treatment of iron deficiency in adults TBD Shield Therapeutics ® Anticoagulant in patients undergoing percutaneous coronary Angiomax RTU (bivalrudin) intervention, including patients with heparin-induced Direct thrombin inhibitor TBD MAIA Pharmaceuticals thrombocytopenia and heparin-induced thrombocytopenia and thrombosis syndrome Baqsimi™ (glucagon) Treatment of severe hypoglycemia in patients with diabetes nasal powder Glucagon analog July 28, 2019 ages 4 years and above Eli Lilly Cuvitru (immune globulin subcutaneous [human], 20% Replacement therapy for primary humoral immunodeficiency solution) 10 mg/50 mL Immunoglobulin July 2, 2019 subcutaneous injection in adult and pediatric patients two years of age and older Baxalta 1 RxHighlights July 2019 Drug name Therapeutic category Indication(s) Launch information Manufacturer(s) Drizalma Sprinkle™ (duloxetine) Treatment of major depressive disorder in adults; generalized Serotonin and norepinephrine anxiety disorder in adults and pediatric -

Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer
The new england journal of medicine Original Article Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer Karim Fizazi, M.D., Neal Shore, M.D., Teuvo L. Tammela, M.D., Ph.D., Albertas Ulys, M.D., Egils Vjaters, M.D., Sergey Polyakov, M.D., Mindaugas Jievaltas, M.D., Murilo Luz, M.D., Boris Alekseev, M.D., Iris Kuss, M.D., Christian Kappeler, Ph.D., Amir Snapir, M.D., Ph.D., Toni Sarapohja, M.Sc., and Matthew R. Smith, M.D., Ph.D., for the ARAMIS Investigators* ABSTRACT BACKGROUND Darolutamide is a structurally unique androgen-receptor antagonist that is under de- From Institut Gustave Roussy, Université velopment for the treatment of prostate cancer. We evaluated the efficacy of darolu- Paris-Sud, Villejuif, France (K.F.); Carolina Urologic Research Center, Myrtle Beach, tamide for delaying metastasis and death in men with nonmetastatic, castration- SC (N.S.); Tampere University Hospital resistant prostate cancer. and University of Tampere, Tampere (T.L.T.), and Orion Pharma, Orion Corporation, METHODS Espoo (A.S., T.S.) — all in Finland; Na- tional Cancer Institute, Vilnius (A.U.), We conducted a randomized, double-blind, placebo-controlled, phase 3 trial involving and Medical Academy, Lithuanian Uni- men with nonmetastatic, castration-resistant prostate cancer and a prostate-specific versity of Health Sciences, Kaunas (M.J.) antigen doubling time of 10 months or less. Patients were randomly assigned in a 2:1 — both in Lithuania; Stradins Clinical University Hospital, Riga, Latvia (E.V.); ratio to receive darolutamide (600 mg [two 300-mg tablets] twice daily) or placebo N.N. Alexandrov National Cancer Center while continuing androgen-deprivation therapy. -

Product Monograph Crestor
PRODUCT MONOGRAPH Pr CRESTOR® rosuvastatin calcium Tablets, 5, 10, 20 and 40 mg LIPID METABOLISM REGULATOR AstraZeneca Canada Inc. Date of Revision: May 14, 2020 1004 Middlegate Road Mississauga, Ontario L4Y 1M4 www.astrazeneca.ca Control No: 235939 CRESTOR® is a registered trademark of the AstraZeneca group of companies. Licensed from Shionogi & Co. Ltd. Osaka, Japan. COPYRIGHT 2003 – 2020 ASTRAZENECA CANADA INC. Page 1 of 46 TABLE OF CONTENTS PART I: HEALTH PROFESSIONAL INFORMATION......................................................3 SUMMARY PRODUCT INFORMATION ..............................................................3 INDICATIONS AND CLINICAL USE....................................................................3 CONTRAINDICATIONS ........................................................................................4 WARNINGS AND PRECAUTIONS .......................................................................5 ADVERSE REACTIONS ...................................................................................... 10 DRUG INTERACTIONS....................................................................................... 16 DOSAGE AND ADMINISTRATION.................................................................... 22 OVERDOSAGE..................................................................................................... 25 ACTION AND CLINICAL PHARMACOLOGY................................................... 25 STORAGE AND STABILITY............................................................................... 27 DOSAGE -

Endosulfan and Endocrine Disruption: Human Risk Characterization
ENDOSULFAN AND ENDOCRINE DISRUPTION: HUMAN RISK CHARACTERIZATION Prepared by Laura M. Plunkett, Ph.D., DABT Integrative Biostrategies, LLC Houston, Texas Prepared for Makhteshim-Agan of North America, Inc. Raleigh, North Carolina June 23, 2008 1 I. Overview of Endocrine Disruption as an Endpoint of Concern in Pesticide Risk Assessment The endocrine system is one of the basic systems in mammalian physiology that is involved in variety of body functions including such functions as growth, development, reproduction, and behavior. The activity of the endocrine system involves endocrine glands which are collections of specialized cells in various tissues of the body that synthesize, store and release substances into the bloodstream. These substances are known as hormones. Hormones act as chemical messengers, traveling through the bloodstream to distant organs and tissues where they can bind to receptors located on those tissues and organs and as a result of receptor binding trigger a physiological response. The major endocrine glands include the pituitary gland, the thyroid gland, the pancreas, the adrenal gland, the testes, and the ovary. “Endocrine disruption”1 is a term that has arisen in human health risk assessment and is defined as the action of an external agent, such as a chemical, to interfere with normal activity of circulating hormones. This “interference” would include altering the synthesis, storage, release, or degradation of hormones, as well as actions at hormone receptors such as stimulating receptor activity (receptor agonist activity) or inhibiting receptor activity (receptor antagonist activity). The potential effects of chemicals, including pesticides, as endocrine disruptors have been a focus of recent regulatory actions. -

Prostate Cancer and Sexual Function
Prostate Cancer and Prostatic Diseases (1998) 1, 179 ±184 ß 1998 Stockton Press All rights reserved 1365±7852/98 $12.00 http://www.stockton-press.co.uk/pc Review Prostate cancer and sexual function RS Kirby,1 A Watson2 and DWW Newling3 1St. George's Hospital, London, UK; 2Watson Biomedical, Maidstone, UK; and 3Academisch Ziekenhuis der Vrije Universiteit, Amsterdam, The Netherlands Erectile dysfunction is a condition affecting 1 in every 10 men. Although its occurrence is related to ageing, illness and its necessary therapy can play a major role. Prostate cancer can lead to erectile dysfunction both psychologically through depression and emotional distress, and physically through therapy for the disease. An international quality of life survey involving 401 patients with prostate cancer was conducted. The objectives of the study were to investigate the patients' understanding of the treatment options they received, to explore the importance of the patient±doctor communication in the treatment of prostate cancer and to see what effect treatment had on patient's sexual function. One of the main ®ndings of the survey was that too little counselling or information on treatment options and their effects on sexual function was provided to patients. Patients themselves felt that psychosexual counselling, in particular, would be helpful. In addition, therapy for prostate cancer appears to have a signi®cant impact on patients' lifestyle and also on their libido, sexual function and activity. Keywords: prostate cancer; erectile dysfunction; quality -

Driving Performance and Delivering New Growth Opportunities
Driving Performance and Delivering New Growth Opportunities /////////// Capital Markets Day London, December 5, 2018 Stefan Oelrich Head of Pharmaceuticals Joerg Moeller Head of Pharmaceuticals R&D Disclaimer Cautionary Statements Regarding Forward-Looking Information This presentation contains forward-looking statements. A forward-looking statement is any statement that does not relate to historical facts and events, but rather reflects Bayer’s current beliefs, expectations and assumptions regarding the future. This applies, in particular, to statements in this presentation on revenue growth, including product introductions and peak sales potential, synergies, especially in relation to the acquisition and integration of Monsanto Company, portfolio adjustments, cost reduction, financial targets and earnings, cash flow generation, deleveraging and other similar statements relating to future performance, including with respect to the markets in which Bayer is active. Although the forward-looking statements contained in this presentation are based upon what Bayer’s management believes are reasonable assumptions, they necessarily involve known and unknown risks and uncertainties that could cause actual results and future events to differ materially from those anticipated in such statements. Forward- looking statements are not guarantees of future performance and undue reliance should not be placed on them. Bayer undertakes no obligation to update forward-looking statements if circumstances or management’s estimates or opinions should change except as required by applicable securities laws. For more information on factors that could cause actual results and future events to differ from those anticipated in forward looking statements, please refer to the factors discussed in Bayer’s public reports which are available on the Bayer website at https://www.investor.bayer.com/en/reports/annual-reports/overview/, including in the Annual Report 2017 under the caption “Report on Future Perspectives and on Opportunities and Risks”. -

Indoor Dust Poses Significant Endocrine Disruptor Risk
9 October 2008 Indoor dust poses significant endocrine disruptor risk The risks from exposure to outdoor pollution or sources like tobacco smoke are well known, but indoor dust can also pose health risks, especially to young children. New evidence shows that indoor dust is highly contaminated by persistent and endocrine-disrupting chemicals, including some chemicals such as polychlorinated biphenyls (PCBs) which have been banned since the 1970s. Endocrine disruptors are substances which disrupt the body’s natural hormonal system. Some endocrine- disrupting chemicals can persist for many years. These include PCBs, polybrominated diphenyl ethers (PBDEs), pyrethroids, dichlorodiphenyltrichloroethane (DDT), chlorodanes and phthalates. These chemicals and others are found in indoor dust, which can be eaten or breathed in. Endocrine disrupters may be responsible for declining sperm counts, genital malformations, cancers and impaired neural development and sexual behaviour. Dust collected from vacuum cleaners used in apartments and a community hall was highly contaminated with endocrine disruptors, in particular phthalates and PBDEs. The levels of PCBs were high enough in some cases to be a health concern, illustrating that these chemicals continue to persist in the environment and pose risks. Although the use of some PBDEs is being phased out in some parts of the US and they have been banned in EU Member States since 2004, this trend may apply to PBDEs as well. PBDEs are flame retardants used on soft furnishings, and so as long as items such as mattresses, carpets and sofas treated with the chemical are used then exposure will continue. Because young children spend a lot of time indoors, often at floor level, and put objects in their mouths, they are particularly at risk from pollutants in indoor dust. -

Connecticut Medicaid
ACNE AGENTS, TOPICAL ‡ ANGIOTENSIN MODULATOR COMBINATIONS ANTICONVULSANTS, CONT. CONNECTICUT MEDICAID (STEP THERAPY CATEGORY) AMLODIPINE / BENAZEPRIL (ORAL) LAMOTRIGINE CHEW DISPERS TAB (not ODT) (ORAL) (DX CODE REQUIRED - DIFFERIN, EPIDUO and RETIN-A) AMLODIPINE / OLMESARTAN (ORAL) LAMOTRIGINE TABLET (IR) (not ER) (ORAL) Preferred Drug List (PDL) ACNE MEDICATION LOTION (BENZOYL PEROXIDE) (TOPICAL)AMLODIPINE / VALSARTAN (ORAL) LEVETIRACETAM SOLUTION, IR TABLET (not ER) (ORAL) • The Connecticut Medicaid Preferred Drug List (PDL) is a BENZOYL PEROXIDE CREAM, WASH (not FOAM) (TOPICAL) OXCARBAZEPINE TABLET (ORAL) listing of prescription products selected by the BENZOYL PEROXIDE 5% and 10% GEL (OTC) (TOPICAL) ANTHELMINTICS PHENOBARBITAL ELIXIR, TABLET (ORAL) Pharmaceutical and Therapeutics Committee as efficacious, BENZOYL PEROXIDE 6% CLEANSER (OTC) (TOPICAL) ALBENDAZOLE TABLET (ORAL) PHENYTOIN CHEW TABLET, SUSPENSION (ORAL) safe and cost effective choices when prescribing for HUSKY CLINDAMYCIN PH 1% PLEGET (TOPICAL) BILTRICIDE TABLET (ORAL) PHENYTOIN SOD EXT CAPSULE (ORAL) A, HUSKY C, HUSKY D, Tuberculosis (TB) and Family CLINDAMYCIN PH 1% SOLUTION (not GEL or LOTION) (TOPICAL)IVERMECTIN TABLET (ORAL) PRIMIDONE (ORAL) Planning (FAMPL) clients. CLINDAMYCIN / BENZOYL PEROXIDE 1.2%-5% (DUAC) (TOPICAL) SABRIL 500 MG POWDER PACK (ORAL) • Preferred or Non-preferred status only applies to DIFFERIN 0.1% CREAM (TOPICAL) (not OTC GEL) (DX CODE REQ.) ANTI-ALLERGENS, ORAL SABRIL TABLET (ORAL) those medications that fall within the drug classes DIFFERIN -

Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-Α
biomedicines Article Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-α Elisa Rossini 1,†, Edoardo Giacopuzzi 2,3,† , Fabrizio Gangemi 4,† , Mariangela Tamburello 1, Deborah Cosentini 5, Andrea Abate 1, Marta Laganà 5, Alfredo Berruti 5 , Salvatore Grisanti 5,‡ and Sandra Sigala 1,*,‡ 1 Section of Pharmacology, Department of Molecular and Translational Medicine, University of Brescia, 25123 Brescia, Italy; [email protected] (E.R.); [email protected] (M.T.); [email protected] (A.A.) 2 Wellcome Centre for Human Genetics, University of Oxford, Oxford OX3 7BN, UK; [email protected] 3 National Institute for Health Research Oxford Biomedical Research Centre, Oxford OX4 2PG, UK 4 Unit of Physics, Department of Molecular and Translational Medicine, University of Brescia, 25123 Brescia, Italy; [email protected] 5 Medical Oncology Unit, Department of Medical and Surgical Specialties, Radiological Sciences, and Public Health, University of Brescia at A.S.S.T. Spedali Civili di Brescia, 25123 Brescia, Italy; [email protected] (D.C.); [email protected] (M.L.); [email protected] (A.B.); [email protected] (S.G.) * Correspondence: [email protected] † These Authors equally contributed to this paper. ‡ These authors are co-senior authors of this paper. Citation: Rossini, E.; Giacopuzzi, E.; Abstract: Mitotane is the cornerstone of medical treatment of adrenocortical carcinoma. Estrogenic- Gangemi, F.; Tamburello, M.; like side effects frequently occur in patients, and previous studies explored the chemical nature Cosentini, D.; Abate, A.; Laganà, M.; of the interaction between estrogen receptor-α (ER-α) and toxic compounds, including the DDD Berruti, A.; Grisanti, S.; Sigala, S. -

HPD V2.2 Created Via HPDC Builder Page 1 of 17 VOLATILE ORGANIC COMPOUND (VOC) CONTENT CERTIFICATIONS and COMPLIANCE See Section 3 for Additional Listings
M2.1 Health Product by Humanscale Declaration v2.2 created via: HPDC Online Builder HPD UNIQUE IDENTIFIER: 21096 CLASSIFICATION: 12 51 00.00 Furnishings: Office Furniture PRODUCT DESCRIPTION: In today’s fast-paced, agile work environment, there’s no time for discomfort. M2.1, part of Humanscale’s revolutionary new monitor arm line, instantly improves the comfort, health and productivity of any workspace. Fully compatible with traditional desks and sit/stand workstations alike, M2.1 meets a variety of configuration needs for lighter monitors up to 15.5 lbs. Featuring innovations like Humanscale’s patented Weight-Compensating Spring Technology and Smart Stop functionality, M2.1 enables the personalization and flexibility needed for today’s evolving workplaces. Section 1: Summary Nested Method / Product Threshold CONTENT INVENTORY Inventory Reporting Format Threshold level Residuals/Impurities All Substances Above the Threshold Indicated Are: Nested Materials Method 100 ppm Residuals/Impurities Characterized Yes Ex/SC Yes No Basic Method 1,000 ppm Considered in 6 of 13 Materials % weight and role provided for all substances. Per GHS SDS Explanation(s) provided Threshold Disclosed Per Other for Residuals/Impurities? Screened Yes Ex/SC Yes No Material Yes No All substances screened using Priority Hazard Lists with Product results disclosed. Identified Yes Ex/SC Yes No One or more substances not disclosed by Name (Specific or Generic) and Identifier and/ or one or more Special Condition did not follow guidance. CONTENT IN DESCENDING ORDER OF QUANTITY Number of Greenscreen BM-4/BM3 contents ... 0 Summary of product contents and results from screening individual chemical Contents highest concern GreenScreen substances against HPD Priority Hazard Lists and the GreenScreen for Safer Benchmark or List translator Score .. -

BPA and Endocrine Disruption
www.bisphenol-a-europe.org Questions and answers about Endocrine Disruption and Bisphenol A There is an ongoing debate and public concern about effects of endocrine disruptors (EDs) on human health and the environment. Endocrine active Endocrine disrupting In that context, the substance Bisphenol A (BPA) is often mentioned substances substances as an example; the public seems to be convinced that “BPA is an interaction with the causing an adverse ED”. This document provides factual background and explains hormone system effect by interacting with the hormonal why Bisphenol A in realistic exposure levels is not a threat to the there are many system endocrine (hormonal) system. naturally occurring endocrine active hormonal contra- substances humans cep tives are a well What is the endocrine system? take in on a daily basis known example as The endocrine system is commonly known as since thousands of they inter fere with the the hormonal system. Hormones are important years, without known hormonal system to negative health effects prevent pregnancy. In messenger-substances produced in the body´s this case the adverse glands. Hormones regulate, among others, metabolic effect is intended and processes, growth, reproduction and development (if desired) limited in in a very complex way. The endocrine system is time naturally prepared to cope with the various external and internal factors influencing the hormonal system. What are endocrine active substances? What are endocrine disrupting substances? Substances that can interact with the hormonal There are some endocrine active substances which system are called endocrine active substances. Such can induce an adverse effect by influencing the substances naturally occur e.g. -

Maximum Androgen Blockade Does Not Confer Additional Survival Benefit to Androgen Suppression in Prostate Cancer
Evid Based Med: first published as 10.1136/ebm.6.1.17 on 1 January 2001. Downloaded from Review: maximum androgen blockade does not confer additional survival benefit to androgen suppression in prostate cancer Prostate Cancer Trialists’ Collaborative Group. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Lancet 2000 Apr 29;355:1491–8. QUESTION: In men with prostate cancer, is maximum androgen blockade (MAB) better than androgen suppression (AS) alone for prolonging survival? Data sources Conclusion Studies were identified by searching computerised data- In men with advanced prostate cancer, the addition of bases, trial registers, meeting abstracts, and reference an antiandrogen to androgen suppression alone does lists and by contacting investigators and pharmaceutical not confer a statistically significant survival benefit at 5 companies. years. Study selection Studies were selected if they were randomised trials that COMMENTARY began before 1991 and compared MAB (AS plus an antiandrogen, such as nilutamide, flutamide, or cyprot- The Prostate Cancer Trialists’ Collaborative Group erone acetate) with AS alone and if the treatment was (PCTCG) study shows that adding a non-steroidal antian- given for >1 year. drogen (NSAA) to androgen suppression produces a statis- tically significant increase in long term survival over AS Data extraction alone. The real question is whether the difference of 2.9% (CI 0.4% to 5.4%) is clinically significant. The answer is, Information was requested for each patient on stage of “probably not.” disease, age at randomisation, date of randomisation, The issue hinges on symptoms and quality of life. Adding treatment allocation, date of last follow up, date of death, an NSAA to medical or surgical castration, thereby achiev- and cause of death.