Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families with Neurodevelopmental Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] Product Data](https://docslib.b-cdn.net/cover/2076/eef1d-mouse-monoclonal-antibody-clone-id-oti4b9-product-data-82076.webp)
EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for CF811676 EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] Product data: Product Type: Primary Antibodies Clone Name: OTI4B9 Applications: IHC, WB Recommended Dilution: WB 1:500~2000, IHC 1:2000 Reactivity: Human, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human EEF1D (NP_115754) produced in E.coli. Formulation: Lyophilized powder (original buffer 1X PBS, pH 7.3, 8% trehalose) Reconstitution Method: For reconstitution, we recommend adding 100uL distilled water to a final antibody concentration of about 1 mg/mL. To use this carrier-free antibody for conjugation experiment, we strongly recommend performing another round of desalting process. (OriGene recommends Zeba Spin Desalting Columns, 7KMWCO from Thermo Scientific) Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Gene Name: Homo sapiens eukaryotic translation elongation factor 1 delta (EEF1D), transcript variant 1, mRNA. Database Link: NP_115754 Entrez Gene 1936 Human P29692 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] – CF811676 Background: This gene encodes a subunit of the elongation factor-1 complex, which is responsible for the enzymatic delivery of aminoacyl tRNAs to the ribosome. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

SEC23IP (NM 007190) Human Recombinant Protein – TP309056
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP309056 SEC23IP (NM_007190) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human SEC23 interacting protein (SEC23IP) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 110.9 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_009121 Locus ID: 11196 UniProt ID: Q9Y6Y8 RefSeq Size: 7306 Cytogenetics: 10q26.11-q26.12 RefSeq ORF: 3000 Synonyms: iPLA1beta; MSTP053; P125; P125A Summary: This gene encodes a member of the phosphatidic acid preferring-phospholipase A1 family. The encoded protein is localized to endoplasmic reticulum exit sites and plays a critical role in ER-Golgi transport as part of the multimeric coat protein II complex. An orthologous gene in frogs is required for normal neural crest cell development, suggesting that this gene may play a role in Waardenburg syndrome neural crest defects. Alternatively spliced transcript variants have been observed for this gene. [provided by RefSeq, Feb 2011] This product is to be used for laboratory only. -

A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2014 A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk Nancy L. Maas University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biology Commons, Cell Biology Commons, and the Molecular Biology Commons Recommended Citation Maas, Nancy L., "A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk" (2014). Publicly Accessible Penn Dissertations. 1354. https://repository.upenn.edu/edissertations/1354 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1354 For more information, please contact [email protected]. A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk Abstract Cells constantly encounter changing environments that challenge the ability to adapt and survive. Signal transduction networks enable cells to appropriately sense and respond to these changes, and are often mediated through the activity of protein kinases. Protein kinases are a class of enzyme responsible for regulating a broad spectrum of cellular functions by transferring phosphate groups from ATP to substrate proteins, thereby altering substrate activity and function. PERK is a resident kinase of the endoplasmic reticulum, and is responsible for sensing perturbations in the protein folding capacity of the ER. When the influx of unfolded, nascent proteins exceeds the folding capacity of the ER, PERK initiates a cascade of signaling events that enable cell adaptation and ER stress resolution. These signaling pathways are not only essential for the survival of normal cells undergoing ER stress, but are also co-opted by tumor cells in order to survive the oxygen and nutrient-restricted conditions of the tumor microenvironment. -

Genetic Variant in 3' Untranslated Region of the Mouse Pycard Gene
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.26.437184; this version posted March 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 3 Title: 4 Genetic Variant in 3’ Untranslated Region of the Mouse Pycard Gene Regulates Inflammasome 5 Activity 6 Running Title: 7 3’UTR SNP in Pycard regulates inflammasome activity 8 Authors: 9 Brian Ritchey1*, Qimin Hai1*, Juying Han1, John Barnard2, Jonathan D. Smith1,3 10 1Department of Cardiovascular & Metabolic Sciences, Lerner Research Institute, Cleveland Clinic, 11 Cleveland, OH 44195 12 2Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 13 44195 14 3Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine of Case Western 15 Reserve University, Cleveland, OH 44195 16 *, These authors contributed equally to this study. 17 Address correspondence to Jonathan D. Smith: email [email protected]; ORCID ID 0000-0002-0415-386X; 18 mailing address: Cleveland Clinic, Box NC-10, 9500 Euclid Avenue, Cleveland, OH 44195, USA. 19 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.26.437184; this version posted March 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 20 Abstract 21 Quantitative trait locus mapping for interleukin-1 release after inflammasome priming and activation 22 was performed on bone marrow-derived macrophages (BMDM) from an AKRxDBA/2 strain intercross. -

Genes with 5' Terminal Oligopyrimidine Tracts Preferentially Escape Global Suppression of Translation by the SARS-Cov-2 NSP1 Protein
Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Genes with 5′ terminal oligopyrimidine tracts preferentially escape global suppression of translation by the SARS-CoV-2 Nsp1 protein Shilpa Raoa, Ian Hoskinsa, Tori Tonna, P. Daniela Garciaa, Hakan Ozadama, Elif Sarinay Cenika, Can Cenika,1 a Department of Molecular Biosciences, University of Texas at Austin, Austin, TX 78712, USA 1Corresponding author: [email protected] Key words: SARS-CoV-2, Nsp1, MeTAFlow, translation, ribosome profiling, RNA-Seq, 5′ TOP, Ribo-Seq, gene expression 1 Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Viruses rely on the host translation machinery to synthesize their own proteins. Consequently, they have evolved varied mechanisms to co-opt host translation for their survival. SARS-CoV-2 relies on a non-structural protein, Nsp1, for shutting down host translation. However, it is currently unknown how viral proteins and host factors critical for viral replication can escape a global shutdown of host translation. Here, using a novel FACS-based assay called MeTAFlow, we report a dose-dependent reduction in both nascent protein synthesis and mRNA abundance in cells expressing Nsp1. We perform RNA-Seq and matched ribosome profiling experiments to identify gene-specific changes both at the mRNA expression and translation level. We discover that a functionally-coherent subset of human genes are preferentially translated in the context of Nsp1 expression. These genes include the translation machinery components, RNA binding proteins, and others important for viral pathogenicity. Importantly, we uncovered a remarkable enrichment of 5′ terminal oligo-pyrimidine (TOP) tracts among preferentially translated genes. -

Early Alterations of RNA Metabolism and Splicing from Adult Corticospinal Neurons In
bioRxiv preprint doi: https://doi.org/10.1101/667733; this version posted June 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Early alterations of RNA metabolism and splicing from adult corticospinal neurons in 2 an ALS mouse model 3 4 Christine Marques1,2, Mathieu Fischer1,3, Céline Keime4, Thibaut Burg1, Aurore Brunet1, 5 Jelena Scekic-Zahirovic1 & Caroline Rouaux1* 6 7 8 9 1Inserm UMR_S 1118, Mécanismes centraux et périphériques de la neurodégénérescence, 10 Faculté de Médecine, Université de Strasbourg, Strasbourg, France. 11 2Current address: Department of Neurobiology, Harvard Medical School, Boston, MA, USA; 12 Department of Neurology, Massachusetts General Hospital, Boston, MA, USA. 13 3Current address: Department of Paediatrics, John Radcliffe Hospital, University of Oxford, 14 Oxford, UK. 15 4Inserm UMR_S 1258, CRNS UMR_S 7104, Université de Strasbourg, IGBMC, Strasbourg, 16 France. 17 18 *Correspondence should be addressed to: C.R. ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/667733; this version posted June 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease clinically defined as the combined degeneration of corticospinal and corticobulbar neurons (CSN), and bulbar and spinal motor neurons (MN). A growing body of evidence points to the motor cortex, where CSN are located, as the potential initiation site of ALS. However, little is known about the spatiotemporal dynamics of CSN degeneration and the molecular pathways involved. -

TITLE PAGE Oxidative Stress and Response to Thymidylate Synthase
Downloaded from molpharm.aspetjournals.org at ASPET Journals on October 2, 2021 -Targeted -Targeted 1 , University of of , University SC K.W.B., South Columbia, (U.O., Carolina, This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. -

Apoptotic Genes As Potential Markers of Metastatic Phenotype in Human Osteosarcoma Cell Lines
17-31 10/12/07 14:53 Page 17 INTERNATIONAL JOURNAL OF ONCOLOGY 32: 17-31, 2008 17 Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines CINZIA ZUCCHINI1, ANNA ROCCHI2, MARIA CRISTINA MANARA2, PAOLA DE SANCTIS1, CRISTINA CAPANNI3, MICHELE BIANCHINI1, PAOLO CARINCI1, KATIA SCOTLANDI2 and LUISA VALVASSORI1 1Dipartimento di Istologia, Embriologia e Biologia Applicata, Università di Bologna, Via Belmeloro 8, 40126 Bologna; 2Laboratorio di Ricerca Oncologica, Istituti Ortopedici Rizzoli; 3IGM-CNR, Unit of Bologna, c/o Istituti Ortopedici Rizzoli, Via di Barbiano 1/10, 40136 Bologna, Italy Received May 29, 2007; Accepted July 19, 2007 Abstract. Metastasis is the most frequent cause of death among malignant primitive bone tumor, usually developing in children patients with osteosarcoma. We have previously demonstrated and adolescents, with a high tendency to metastasize (2). in independent experiments that the forced expression of Metastases in osteosarcoma patients spread through peripheral L/B/K ALP and CD99 in U-2 OS osteosarcoma cell lines blood very early and colonize primarily the lung, and later markedly reduces the metastatic ability of these cancer cells. other skeleton districts (3). Since disseminated hidden micro- This behavior makes these cell lines a useful model to assess metastases are present in 80-90% of OS patients at the time the intersection of multiple and independent gene expression of diagnosis, the identification of markers of invasiveness signatures concerning the biological problem of dissemination. and metastasis forms a target of paramount importance in With the aim to characterize a common transcriptional profile planning the treatment of osteosarcoma lesions and enhancing reflecting the essential features of metastatic behavior, we the prognosis. -

Role of Cyclin-Dependent Kinase 1 in Translational Regulation in the M-Phase
cells Review Role of Cyclin-Dependent Kinase 1 in Translational Regulation in the M-Phase Jaroslav Kalous *, Denisa Jansová and Andrej Šušor Institute of Animal Physiology and Genetics, Academy of Sciences of the Czech Republic, Rumburska 89, 27721 Libechov, Czech Republic; [email protected] (D.J.); [email protected] (A.Š.) * Correspondence: [email protected] Received: 28 April 2020; Accepted: 24 June 2020; Published: 27 June 2020 Abstract: Cyclin dependent kinase 1 (CDK1) has been primarily identified as a key cell cycle regulator in both mitosis and meiosis. Recently, an extramitotic function of CDK1 emerged when evidence was found that CDK1 is involved in many cellular events that are essential for cell proliferation and survival. In this review we summarize the involvement of CDK1 in the initiation and elongation steps of protein synthesis in the cell. During its activation, CDK1 influences the initiation of protein synthesis, promotes the activity of specific translational initiation factors and affects the functioning of a subset of elongation factors. Our review provides insights into gene expression regulation during the transcriptionally silent M-phase and describes quantitative and qualitative translational changes based on the extramitotic role of the cell cycle master regulator CDK1 to optimize temporal synthesis of proteins to sustain the division-related processes: mitosis and cytokinesis. Keywords: CDK1; 4E-BP1; mTOR; mRNA; translation; M-phase 1. Introduction 1.1. Cyclin Dependent Kinase 1 (CDK1) Is a Subunit of the M Phase-Promoting Factor (MPF) CDK1, a serine/threonine kinase, is a catalytic subunit of the M phase-promoting factor (MPF) complex which is essential for cell cycle control during the G1-S and G2-M phase transitions of eukaryotic cells. -
Type of the Paper (Article
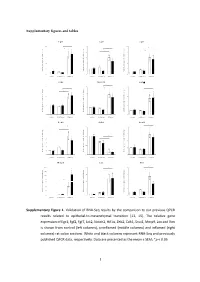
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

SEC23IP (NM 007190) Human Tagged ORF Clone – RG209056
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG209056 SEC23IP (NM_007190) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: SEC23IP (NM_007190) Human Tagged ORF Clone Tag: TurboGFP Symbol: SEC23IP Synonyms: iPLA1beta; MSTP053; P125; P125A Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 SEC23IP (NM_007190) Human Tagged ORF Clone – RG209056 ORF Nucleotide >RG209056 representing NM_007190 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCCGAGAGAAAACCTAACGGTGGCAGCGGCGGCGCCTCCACTTCCTCATCGGGCACTAACTTACTTT TCTCCTCCTCGGCCACGGAGTTCAGCTTCAATGTGCCCTTCATCCCAGTCACCCAGGCCTCCGCTTCTCC GGCCTCCCTGCTCTTACCGGGAGAGGATTCCACAGATGTTGGTGAGGAGGACAGCTTCCTTGGTCAGACT TCTATTCACACATCTGCCCCACAGACATTTAGTTACTTCTCTCAGGTATCAAGCAGCAGTGATCCTTTTG GGAATATTGGACAGTCACCATTAACAACTGCAGCAACCTCAGTTGGACAATCAGGATTCCCCAAGCCCCT GACTGCTCTCCCTTTTACAACTGGATCCCAAGATGTCTCGAATGCATTTTCACCATCCATTTCGAAGGCT CAACCTGGTGCTCCACCTTCCTCACTGATGGGAATAAATTCTTATCTGCCTTCTCAGCCAAGTAGTCTCC CTCCTTCATATTTTGGGAACCAACCCCAAGGAATTCCCCAACCAGGATACAATCCATATCGCCATACCCC TGGCAGCAGCAGGGCTAATCCTTACATTGCACCACCCCAGCTGCAGCAGTGCCAAACACCAGGCCCTCCT