Galapagos NV R&D Update on June 20, 2017 / 12:00PM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GALAPAGOS NV (Name of Issuer)
SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 SCHEDULE 13D Under the Securities Exchange Act of 1934 (Amendment No. )* GALAPAGOS NV (Name of Issuer) Ordinary Shares, no par value (Title of Class of Securities) 36315X101 (CUSIP Number) Gilead Sciences, Inc. 333 Lakeside Drive Foster City, California, 94404 650-574-3000 (Name, Address and Telephone Number of Person Authorized to Receive Notices and Communications) July 14, 2019 (Date of Event Which Requires Filing of this Statement) If the filing person has previously filed a statement on Schedule 13G to report the acquisition that is the subject of this Schedule 13D, and is filing this schedule because of §§240.13d-1(e), 240.13d-1(f) or 240.13d-1(g), check the following box. x Note: Schedules filed in paper format shall include a signed original and five copies of the schedule, including all exhibits. See Rule 13d- 7(b) for other parties to whom copies are to be sent. *The remainder of this cover page shall be filled out for a reporting person’s initial filing on this form with respect to the subject class of securities, and for any subsequent amendment containing information which would alter disclosures provided in a prior cover page. The information required on the remainder of this cover page shall not be deemed to be “filed” for the purpose of Section 18 of the Securities Exchange Act of 1934 (the “Act”) or otherwise subject to the liabilities of that section of the Act but shall be subject to all other provisions of the Act (however, see the Notes). -

Additional Details for the Item 4 of the Extraordinary General Meeting of Euronext N.V
CONTACT - Investor Relations: +33.1.70.48.24.17 ADDITIONAL DETAILS FOR THE ITEM 4 OF THE EXTRAORDINARY GENERAL MEETING OF EURONEXT N.V. ON 8 OCTOBER 2019 To ensure clear and sufficient understanding of the items submitted for shareholder approval at the Extraordinary General Meeting of Euronext N.V. on 8 October 2019, the following additional information regarding Item 4 – Proposal to amend the remuneration policy is disclosed. The reader is invited to refer to the full explanatory note document available on https://www.euronext.com/en/investor-relations/financial- calendar/egm-8102019, where the full remuneration policy is made available in Appendix 3. Refering to the notes related to Proposal 4, it is mentioned that “in accordance with the Remuneration policy, a benchmark analysis has been conducted against four different peer groups to assess compensation levels of the CEO. First two reference markets consist of European headquartered companies of comparable size and scope, primarily active in the Finance & IT industries (>20 companies) and relevant direct competitors (>20 companies). To complement these two peer group, an analysis was performed against two local markets on equivalent position (France where the CEO is based (>20 companies, and the Netherlands/AEX (>15 companies). This analysis shows that the current target total compensation package is positioned below European Market, significantly below the median of the French Market and the lower quartile of the Direct Competitors and slightly lower than the Dutch Market.” Furthermore, -

Retirement Strategy Fund 2060 Description Plan 3S DCP & JRA
Retirement Strategy Fund 2060 June 30, 2020 Note: Numbers may not always add up due to rounding. % Invested For Each Plan Description Plan 3s DCP & JRA ACTIVIA PROPERTIES INC REIT 0.0137% 0.0137% AEON REIT INVESTMENT CORP REIT 0.0195% 0.0195% ALEXANDER + BALDWIN INC REIT 0.0118% 0.0118% ALEXANDRIA REAL ESTATE EQUIT REIT USD.01 0.0585% 0.0585% ALLIANCEBERNSTEIN GOVT STIF SSC FUND 64BA AGIS 587 0.0329% 0.0329% ALLIED PROPERTIES REAL ESTAT REIT 0.0219% 0.0219% AMERICAN CAMPUS COMMUNITIES REIT USD.01 0.0277% 0.0277% AMERICAN HOMES 4 RENT A REIT USD.01 0.0396% 0.0396% AMERICOLD REALTY TRUST REIT USD.01 0.0427% 0.0427% ARMADA HOFFLER PROPERTIES IN REIT USD.01 0.0124% 0.0124% AROUNDTOWN SA COMMON STOCK EUR.01 0.0248% 0.0248% ASSURA PLC REIT GBP.1 0.0319% 0.0319% AUSTRALIAN DOLLAR 0.0061% 0.0061% AZRIELI GROUP LTD COMMON STOCK ILS.1 0.0101% 0.0101% BLUEROCK RESIDENTIAL GROWTH REIT USD.01 0.0102% 0.0102% BOSTON PROPERTIES INC REIT USD.01 0.0580% 0.0580% BRAZILIAN REAL 0.0000% 0.0000% BRIXMOR PROPERTY GROUP INC REIT USD.01 0.0418% 0.0418% CA IMMOBILIEN ANLAGEN AG COMMON STOCK 0.0191% 0.0191% CAMDEN PROPERTY TRUST REIT USD.01 0.0394% 0.0394% CANADIAN DOLLAR 0.0005% 0.0005% CAPITALAND COMMERCIAL TRUST REIT 0.0228% 0.0228% CIFI HOLDINGS GROUP CO LTD COMMON STOCK HKD.1 0.0105% 0.0105% CITY DEVELOPMENTS LTD COMMON STOCK 0.0129% 0.0129% CK ASSET HOLDINGS LTD COMMON STOCK HKD1.0 0.0378% 0.0378% COMFORIA RESIDENTIAL REIT IN REIT 0.0328% 0.0328% COUSINS PROPERTIES INC REIT USD1.0 0.0403% 0.0403% CUBESMART REIT USD.01 0.0359% 0.0359% DAIWA OFFICE INVESTMENT -

Aktiebeholdning Pr. 05.05.2021 Oversigt Over Investeringer I
Aktiebeholdning pr. 05.05.2021 Oversigt over investeringer i børsnoterede aktier som PenSam Pension har foretaget enten direkte eller gennem investeringsforeninger. Specifikation af beholdningen af børsnoterede aktier Land Selskab/aktie Branche Markedsværdi i kr. Argentina MercadoLibre Inc Consumer Discretionary 49.439.459 YPF SA Energy 1.195.299 Australia Afterpay Ltd Information Technology 7.516.309 AMP Ltd Financials 1.126.955 Ampol Ltd Energy 281.797 APA Group Utilities 2.536.557 Aristocrat Leisure Ltd Consumer Discretionary 27.487.387 ASX Ltd Financials 4.697.579 Aurizon Holdings Ltd Industrials 9.540.418 AusNet Services Utilities 612.886 Australia & New Zealand Banking Group Ltd Financials 21.696.049 Brambles Ltd Industrials 28.287.023 Coca-Cola Amatil Ltd Consumer Staples 1.445.126 Cochlear Ltd Health Care 2.148.881 Commonwealth Bank of Australia Financials 51.943.000 Computershare Ltd Information Technology 12.404.653 CSL Ltd Health Care 83.573.855 Dexus Real Estate 9.005.111 Evolution Mining Ltd Materials 1.690.740 Fortescue Metals Group Ltd Materials 19.528.992 Goodman Group Real Estate 10.217.406 GPT Group/The Real Estate 2.889.633 Insurance Australia Group Ltd Financials 3.939.805 Lendlease Corp Ltd Real Estate 2.589.152 Magellan Financial Group Ltd Financials 2.093.797 Medibank Pvt Ltd Financials 2.869.133 Mirvac Group Real Estate 3.496.016 National Australia Bank Ltd Financials 25.112.465 Newcrest Mining Ltd Materials 15.692.715 Northern Star Resources Ltd Materials 255.676 Oil Search Ltd Energy 460.619 QBE Insurance Group -

Fiscal Quarter-End Holdings (Pdf)
Quarterly Schedules of Portfolio Holdings International & Global Funds July 31, 2020 Retirement Institutional Administrative Investor Class Class Class Class Harbor Diversified International All Cap Fund HNIDX HAIDX HRIDX HIIDX Harbor Emerging Markets Equity Fund HNEMX HAEMX HREMX HIEEX Harbor Focused International Fund HNFRX HNFSX HNFDX HNFIX Harbor Global Leaders Fund HNGIX HGGAX HRGAX HGGIX Harbor International Fund HNINX HAINX HRINX HIINX Harbor International Growth Fund HNGFX HAIGX HRIGX HIIGX Harbor International Small Cap Fund HNISX HAISX HRISX HIISX Harbor Overseas Fund HAORX HAOSX HAOAX HAONX Table of Contents Portfolios of Investments HARBOR DIVERSIFIED INTERNATIONAL ALL CAP FUND. ..... 1 HARBOR EMERGING MARKETS EQUITY FUND . .......... 8 HARBOR FOCUSED INTERNATIONAL FUND. ................................. 11 HARBOR GLOBAL LEADERS FUND. ................. 13 HARBOR INTERNATIONAL FUND . 15 HARBOR INTERNATIONAL GROWTH FUND . ........................ 21 HARBOR INTERNATIONAL SMALL CAP FUND. ................. 24 HARBOR OVERSEAS FUND . ............................................ 26 Notes to Portfolios of Investments ..................................... 31 Harbor Diversified International All Cap Fund PORTFOLIO OF INVESTMENTS—July 31, 2020 (Unaudited) Value, Cost, and Principal Amounts in Thousands COMMON STOCKS—96.4% COMMON STOCKS—Continued Shares Value Shares Value AEROSPACE & DEFENSE—0.7% BANKS—Continued 28,553 Airbus SE (France)* .............................. $ 2,090 236,142 Svenska Handelsbanken AB (Sweden) ............... $ -
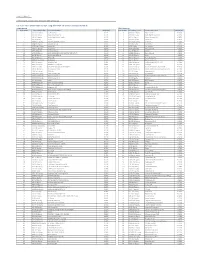
Citi Pure Price Momentum Europe Long-Short Net TR Series II Index
Date: 23-May-21 Index Weights as of monthly rebalance date 12-May-21 Citi Pure Price Momentum Europe Long-Short Net TR Series II Index (CIISPME2) Long Exposure Short Exposure Constituent Bloomberg Ticker Constituent Name Weight(%) Constituent Bloomberg Ticker Constituent Name Weight(%) 1 1COV GY Equity Covestro AG 0.41% 1 ABBN SE Equity ABB LTD-REG -0.16% 2 AAL LN Equity Anglo American Plc 0.27% 2 ABN NA Equity ABN AMRO Group NV -0.29% 3 ABF LN Equity Associated British Foods 0.74% 3 ACA FP Equity Credit Agricole SA -0.89% 4 AC FP Equity Accor 0.32% 4 ADS GY Equity Adidas AG -0.96% 5 ADEN SE Equity ADECCO GROUP AG-REG 0.42% 5 AENA SQ Equity AENA SA -0.18% 6 ADM LN Equity Admiral Group 1.34% 6 AD NA Equity Ahold NV -0.87% 7 ADYEN NA Equity Adyen NV 0.25% 7 AI FP Equity Air Liquide -0.44% 8 AGN NA Equity Aegon NV 0.26% 8 ALFA SS Equity Alfa Laval AB -0.62% 9 AHT LN Equity Ashtead Group 2.81% 9 ALO FP Equity Alstom -1.02% 10 AKZA NA Equity AKZO NOBEL NV EUR0.50(POST REV SPLIT) 1.11% 10 ASSAB SS Equity Assa Abloy B -0.44% 11 ALC SE Equity ALCON AG CHF0.04 0.20% 11 ATO FP Equity AtoS -2.15% 12 ALV GY Equity Allianz SE 0.64% 12 AZN LN Equity AstraZeneca Plc -1.00% 13 AMBUB DC Equity Ambu A/S 0.10% 13 BA/ LN Equity BAE Systems Plc -1.05% 14 AMP IM Equity Amplifon SpA 0.58% 14 BAER SE Equity JULIUS BAER GROUP LTD -0.06% 15 AMS SQ Equity Amadeus IT Hldg SA 0.86% 15 BARC LN Equity Barclays -0.39% 16 VNA GY Equity Deutsche Annington Immobilien 0.03% 16 BATS LN Equity British American Tobacco Plc -0.47% 17 ANTO LN Equity Antofagasta Hldgs 0.44% 17 -
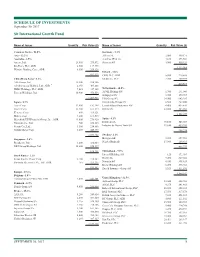
This Is the Message
SCHEDULE OF INVESTMENTS September 30, 2017 Sit International Growth Fund Name of Issuer Quantity Fair Value ($) Name of Issuer Quantity Fair Value ($) Common Stocks - 96.0% Germany - 5.3% Asia - 22.2% Allianz SE 2,000 449,171 Australia - 2.5% Aurelius SE & Co. 4,160 273,564 Amcor, Ltd. 23,400 279,872 Siemens AG 3,900 550,324 Rio Tinto, PLC, ADR 2,500 117,975 1,273,059 Westpac Banking Corp., ADR 8,300 209,326 Ireland - 1.8% 607,173 CRH, PLC, ADR 5,800 219,588 China/Hong Kong - 6.2% Medtronic, PLC 2,700 209,979 AIA Group, Ltd. 32,200 238,386 429,567 Alibaba Group Holding, Ltd., ADR * 2,350 405,868 HSBC Holdings, PLC, ADR 7,025 347,105 Netherlands - 10.5% Tencent Holdings, Ltd. 10,900 476,556 ASML Holding NV 1,700 291,040 Galapagos NV * 3,725 379,717 1,467,915 ING Groep NV 34,900 643,285 Japan - 8.7% Koninklijke Philips NV 6,500 267,800 Asics Corp. 12,900 192,398 LyondellBasell Industries NV 4,400 435,820 Daicel Corp. 13,400 161,574 RELX NV 22,800 485,033 Keyence Corp. 600 319,121 2,502,695 Makita Corp. 4,200 169,515 Mitsubishi UFJ Financial Group, Inc., ADR 43,000 276,920 Spain - 4.1% Nintendo Co., Ltd. 700 258,113 Iberdrola SA 70,100 545,069 Secom Co., Ltd. 3,500 254,886 Industria de Diseno Textil SA 11,650 439,205 Suzuki Motor Corp. 8,400 440,875 984,274 2,073,402 Sweden - 2.1% Singapore - 2.6% Hexagon AB 5,200 257,962 Broadcom, Ltd. -

Annual Report 2018 CONTENTS
Think big Annual Report 2018 CONTENTS Contents The Galapagos group CSR report Letter from the management ........................... 4 Our commitment .............................................. 68 At a glance ........................................................... 8 Improving people’s lives .................................. 69 Strategy ............................................................. 10 Diversity and Human Capital Management .. 71 Think big ............................................................ 13 Business ethics ................................................. 74 Going concern statement ............................... 21 Environment, Health, and Safety .................... 77 Risk management and internal control ......... 21 The Galapagos share ....................................... 23 Corporate governance Overview of Galapagos NV ............................. 25 Policies ............................................................... 80 Disclaimer and other information ................. 27 Board of directors ............................................ 80 Committees ....................................................... 84 R&D Share capital and shares ................................. 87 The Galapagos pipeline ................................... 30 Shareholders ..................................................... 91 Target discovery platform ............................... 31 Remuneration report ....................................... 92 Filgotinib ........................................................... -

AI Powered International Equity ETF Schedule of Investments August 31, 2020 (Unaudited)
AI Powered International Equity ETF Schedule of Investments August 31, 2020 (Unaudited) Shares Security Description Value COMMON STOCKS - 99.4% Argentina - 1.1% 360 Globant SA (a) $ 63,929 Australia - 3.3% 512 Atlassian Corporation plc - Class A (a) 98,182 700 Australia & New Zealand Banking Group, Ltd. - ADR 9,463 3,684 Mesoblast, Ltd. - ADR (a) 71,323 1,568 National Australia Bank, Ltd. - ADR 10,396 189,364 Belgium - 1.5% 233 Galapagos NV - ADR (a) 31,026 1,375 Materialise NV - ADR (a) 55,908 86,934 Canada - 38.0% 7,200 Alexco Resource Corporation (a) 22,248 1,089 Algonquin Power & Utilities Corporation 15,061 914 Aurinia Pharmaceuticals, Inc. (a) 13,555 12,605 Auryn Resources, Inc. (a) 24,580 1,140 B2Gold Corporation 7,684 2,052 Ballard Power Systems, Inc. (a) 34,063 1,650 Barrick Gold Corporation 48,923 132 BCE, Inc. 5,677 4,774 BlackBerry, Ltd. (a) 24,920 748 Brookfield Asset Management, Inc. - Class A 25,238 1,049 CAE, Inc. (a) 16,585 376 Canada Goose Holdings, Inc. (a) 9,208 462 Canadian National Railway Company 48,316 1,963 Canadian Natural Resources, Ltd. 38,612 146 Canadian Pacific Railway, Ltd. 43,169 1,491 Canadian Solar, Inc. (a) 48,443 1,635 Canopy Growth Corporation (a) 26,963 840 Canopy Growth Corporation (a) 13,877 2,220 Cascades, Inc. 23,607 2,744 Celestica, Inc. (a) 21,815 11 Constellation Software, Inc. 12,734 1,064 Descartes Systems Group, Inc. (a) 64,851 2,039 Dollarama, Inc. 79,600 1,597 Eldorado Gold Corporation (a) 18,238 13,136 EMX Royalty Corporation (a) 38,357 17,174 Enerplus Corporation 45,511 1,337 Fortis, Inc. -
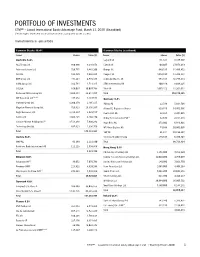
Portfolio of Investments
PORTFOLIO OF INVESTMENTS CTIVP® – Lazard International Equity Advantage Fund, March 31, 2020 (Unaudited) (Percentages represent value of investments compared to net assets) Investments in securities Common Stocks 96.4% Common Stocks (continued) Issuer Shares Value ($) Issuer Shares Value ($) Australia 6.4% Legrand SA 131,321 8,375,908 AGL Energy Ltd. 880,686 9,216,678 L’Oreal SA 104,607 27,073,419 Aristocrat Leisure Ltd. 728,775 9,443,206 Orange SA 949,535 11,496,452 ASX Ltd. 162,820 7,642,885 Peugeot SA 1,014,598 13,209,331 BHP Group Ltd. 471,264 8,549,345 Schneider Electric SE 553,434 46,777,833 CIMIC Group Ltd. 332,784 4,713,315 STMicroelectronics NV 400,446 8,608,225 CSL Ltd. 166,037 30,097,798 Total SA 1,095,152 41,264,011 Fortescue Metals Group Ltd. 4,669,354 28,614,857 Total 209,274,345 (a),(b) IDP Education Ltd. 447,152 3,487,548 Germany 4.2% Inghams Group Ltd. 1,200,179 2,405,115 Adidas AG 22,586 5,014,708 Magellan Financial Group Ltd. 729,823 19,356,167 Allianz SE, Registered Shares 319,746 54,451,936 Regis Resources Ltd. 1,318,187 2,929,557 Continental AG 32,324 2,304,645 Santos Ltd. 4,606,425 9,460,470 Dialog Semiconductor PLC(c) 82,878 2,154,248 (c) Saracen Mineral Holdings Ltd. 3,513,246 7,900,242 Hugo Boss AG 273,602 6,848,821 Technology One Ltd. 897,813 4,336,750 MTU Aero Engines AG 74,698 10,801,095 Total 148,153,933 SAP SE 91,344 10,198,367 Austria 0.2% Siemens Healthineers AG 179,517 6,939,504 OMV AG 81,189 2,221,890 Total 98,713,324 Raiffeisen Bank International AG 111,223 1,599,856 Hong Kong 3.9% Total 3,821,746 CK Hutchison Holdings Ltd. -

Investment Holdings As of June 30, 2019
Investment Holdings As of June 30, 2019 Montana Board of Investments | Portfolio as of June 30, 2019 Transparency of the Montana Investment Holdings The Montana Board of Investment’s holdings file is a comprehensive listing of all manager funds, separately managed and commingled, and aggregated security positions. Securities are organized across common categories: Pension Pool, Asset Class, Manager Fund, Aggregated Individual Holdings, and Non-Pension Pools. Market values shown are in U.S. dollars. The market values shown in this document are for the individual investment holdings only and do not include any information on accounts for receivables or payables. Aggregated Individual Holdings represent securities held at our custodian bank and individual commingled accounts. The Investment Holdings Report is unaudited and may be subject to change. The audited Unified Investment Program Financial Statements, prepared on a June 30th fiscal year-end basis, will be made available once the Legislative Audit Division issues the Audit Opinion. Once issued, the Legislative Audit Division will have the Audit Opinion available online at https://www.leg.mt.gov/publications/audit/agency-search-report and the complete audited financial statements will also be available on the Board’s website http://investmentmt.com/AnnualReportsAudits. Additional information can be found at www.investmentmt.com Montana Board of Investments | Portfolio as of June 30, 2019 2 Table of Contents Consolidated Asset Pension Pool (CAPP) 4 CAPP - Domestic Equities 5 CAPP - International -

R&D Update 2016 Galapagos
137300906113 Galapagos NV (R&D Update 2016) June 15, 2016 C: Elizabeth Goodwin; Galapagos NV; Head of IR C: Bart Filius; Galapagos NV; CFO C: Piet Wigerinck; Galapagos NV; CSO P: Peter Welford; Jefferies; Analyst P: Matthew Harrison; Morgan Stanley; Analyst P: Jan de Kerpel; KBC Securities; Analyst P: Phil Nadeau; Cowen; Analyst P: Hugo Solvet; Bryan, Garnier & Co.; Analyst P: Vamil Divan; Credit Suisse; Analyst P: Jorn Koch; Rabobank; Analyst P: Amanda Weyerbacher; inThought Research; Analyst +++ presentation Elizabeth Goodwin^ Welcome to all of you. We have a large group here at the Yale Club in New York and we have a large group of folks who are dialed in now looking on our webcast as well. So we’re going to try and accommodate all these audiences today, and so I appreciate your patience with some of the technical details. I’d like to ask all the folks in the audience to mute their telephones at this time if they’ve not already done so. Wifi codes are available at your table on the card. I’d like for those who are listening in to the webcast to dial in on the following number, 32-2404-0662 and there’s a code; 207- 496 if you’d like to ask any questions at the end of the presentation. We’re going to have a presentation of one-and-a-half hours and we’ll have about a 30 minute session for Q&A as well. Now, during this presentation, we will be making forward-looking statements. Actual results may differ materially from these statements.