A Novel Nonsense Mutation in MYO15A Is
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

An Uncommon Clinical Presentation of Relapsing Dilated Cardiomyopathy with Identification of Sequence Variations in MYNPC3, KCNH2 and Mitochondrial Trna Cysteine M
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by George Washington University: Health Sciences Research Commons (HSRC) Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 6-2015 An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine M. J. Guillen Sacoto Kimberly A. Chapman George Washington University D. Heath M. B. Seprish Dina Zand George Washington University Follow this and additional works at: http://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Pediatrics Commons Recommended Citation Guillen Sacoto, M.J., Chapman, K.A., Heath, D., Seprish, M.B., Zand, D.J. (2015). An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine. Molecular Genetics and Metabolism Reports, 3, 47-54. doi:10.1016/j.ymgmr.2015.03.007 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. Molecular Genetics and Metabolism Reports 3 (2015) 47–54 Contents lists available at ScienceDirect Molecular Genetics and Metabolism Reports journal homepage: http://www.journals.elsevier.com/molecular-genetics-and- metabolism-reports/ Case Report An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine Maria J. Guillen Sacoto a,1, Kimberly A. -

Novel Myosin Mutations for Hereditary Hearing Loss Revealed by Targeted Genomic Capture and Massively Parallel Sequencing
European Journal of Human Genetics (2014) 22, 768–775 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg ARTICLE Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing Zippora Brownstein1,6, Amal Abu-Rayyan2,6, Daphne Karfunkel-Doron1, Serena Sirigu3, Bella Davidov4, Mordechai Shohat1,4, Moshe Frydman1,5, Anne Houdusse3, Moien Kanaan2 and Karen B Avraham*,1 Hereditary hearing loss is genetically heterogeneous, with a large number of genes and mutations contributing to this sensory, often monogenic, disease. This number, as well as large size, precludes comprehensive genetic diagnosis of all known deafness genes. A combination of targeted genomic capture and massively parallel sequencing (MPS), also referred to as next-generation sequencing, was applied to determine the deafness-causing genes in hearing-impaired individuals from Israeli Jewish and Palestinian Arab families. Among the mutations detected, we identified nine novel mutations in the genes encoding myosin VI, myosin VIIA and myosin XVA, doubling the number of myosin mutations in the Middle East. Myosin VI mutations were identified in this population for the first time. Modeling of the mutations provided predicted mechanisms for the damage they inflict in the molecular motors, leading to impaired function and thus deafness. The myosin mutations span all regions of these molecular motors, leading to a wide range of hearing phenotypes, reinforcing the key role of this family of proteins in auditory function. This study demonstrates that multiple mutations responsible for hearing loss can be identified in a relatively straightforward manner by targeted-gene MPS technology and concludes that this is the optimal genetic diagnostic approach for identification of mutations responsible for hearing loss. -

Using Large Sequencing Data Sets to Refine Intragenic Disease Regions and Prioritize Clinical Variant Interpretation
ORIGINAL RESEARCH ARTICLE © American College of Medical Genetics and Genomics Using large sequencing data sets to refine intragenic disease regions and prioritize clinical variant interpretation Sami S. Amr, PhD1,2, Saeed H. Al Turki, PhD1, Matthew Lebo, PhD1,2, Mahdi Sarmady, PhD3, Heidi L. Rehm, PhD1,2 and Ahmad N. Abou Tayoun, PhD3 Purpose: Classification of novel variants is a major challenge fac- commonly impacted in symptomatic individuals. Our data show ing the widespread adoption of comprehensive clinical genomic that a significant number of exons and domains in genes strongly sequencing and the field of personalized medicine in general. This is associated with disease can be defined as disease-sensitive or disease- largely because most novel variants do not have functional, genetic, tolerant, leading to potential reclassification of at least 26% (450 out or population data to support their clinical classification. of 1,742) of variants of uncertain clinical significance in the 132 genes. Methods: To improve variant interpretation, we leveraged the Exome Conclusion: This approach leverages domain functional annotation Aggregation Consortium (ExAC) data set (N = ~60,000) as well as and associated disease in each gene to prioritize candidate disease 7,000 clinically curated variants in 132 genes identified in more than variants, increasing the sensitivity and specificity of novel variant 11,000 probands clinically tested for cardiomyopathies, rasopathies, assessment within these genes. hearing loss, or connective tissue disorders -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Genome-Wide Identification, Characterization and Expression
G C A T T A C G G C A T genes Article Genome-Wide Identification, Characterization and Expression Profiling of myosin Family Genes in Sebastes schlegelii Chaofan Jin 1, Mengya Wang 1,2, Weihao Song 1 , Xiangfu Kong 1, Fengyan Zhang 1, Quanqi Zhang 1,2,3 and Yan He 1,2,* 1 MOE Key Laboratory of Molecular Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China; [email protected] (C.J.); [email protected] (M.W.); [email protected] (W.S.); [email protected] (X.K.); [email protected] (F.Z.); [email protected] (Q.Z.) 2 Laboratory of Tropical Marine Germplasm Resources and Breeding Engineering, Sanya Oceanographic Institution, Ocean University of China, Sanya 572000, China 3 Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266003, China * Correspondence: [email protected]; Tel.: +86-0532-82031986 Abstract: Myosins are important eukaryotic motor proteins that bind actin and utilize the energy of ATP hydrolysis to perform a broad range of functions such as muscle contraction, cell migration, cytokinesis, and intracellular trafficking. However, the characterization and function of myosin is poorly studied in teleost fish. In this study, we identified 60 myosin family genes in a marine teleost, black rockfish (Sebastes schlegelii), and further characterized their expression patterns. myosin showed divergent expression patterns in adult tissues, indicating they are involved in different types and Citation: Jin, C.; Wang, M.; Song, W.; compositions of muscle fibers. Among 12 subfamilies, S. schlegelii myo2 subfamily was significantly Kong, X.; Zhang, F.; Zhang, Q.; He, Y. -

Novel Gene Discovery in Primary Ciliary Dyskinesia
Novel Gene Discovery in Primary Ciliary Dyskinesia Mahmoud Raafat Fassad Genetics and Genomic Medicine Programme Great Ormond Street Institute of Child Health University College London A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy University College London 1 Declaration I, Mahmoud Raafat Fassad, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract Primary Ciliary Dyskinesia (PCD) is one of the ‘ciliopathies’, genetic disorders affecting either cilia structure or function. PCD is a rare recessive disease caused by defective motile cilia. Affected individuals manifest with neonatal respiratory distress, chronic wet cough, upper respiratory tract problems, progressive lung disease resulting in bronchiectasis, laterality problems including heart defects and adult infertility. Early diagnosis and management are essential for better respiratory disease prognosis. PCD is a highly genetically heterogeneous disorder with causal mutations identified in 36 genes that account for the disease in about 70% of PCD cases, suggesting that additional genes remain to be discovered. Targeted next generation sequencing was used for genetic screening of a cohort of patients with confirmed or suggestive PCD diagnosis. The use of multi-gene panel sequencing yielded a high diagnostic output (> 70%) with mutations identified in known PCD genes. Over half of these mutations were novel alleles, expanding the mutation spectrum in PCD genes. The inclusion of patients from various ethnic backgrounds revealed a striking impact of ethnicity on the composition of disease alleles uncovering a significant genetic stratification of PCD in different populations. -

SARS-Cov-2 Infection of Human Ipsc-Derived Cardiac Cells Predicts Novel Cytopathic Features in Hearts of COVID-19 Patients
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.25.265561; this version posted September 12, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. SARS-CoV-2 infection of human iPSC-derived cardiac cells predicts novel cytopathic features in hearts of COVID-19 patients Juan A. Pérez-Bermejo*1, Serah Kang*1, Sarah J. Rockwood*1, Camille R. Simoneau*1,2, David A. Joy1,3, Gokul N. Ramadoss1,2, Ana C. Silva1, Will R. Flanigan1,3, Huihui Li1, Ken Nakamura1,4, Jeffrey D. Whitman5, Melanie Ott†,1, Bruce R. Conklin†,1,6,7,8, Todd C. McDevitt†1,9 * These authors contributed equally to this work. † Co-corresponding authors. 1 Gladstone Institutes, San Francisco, CA 2 Biomedical Sciences PhD Program, University of California, San Francisco, CA 3 UC Berkeley UCSF Joint Program in Bioengineering, Berkeley, CA 4 UCSF Department of Neurology, San Francisco, CA 5 UCSF Department of Laboratory Medicine, San Francisco, CA 6 Innovative Genomics Institute, Berkeley, CA 7 UCSF Department of Ophthalmology, San Francisco, CA 8 UCSF Department of Medicine, San Francisco, CA 9 UCSF Department of Bioengineering and Therapeutic Sciences, San Francisco, CA ABSTRACT Although COVID-19 causes cardiac dysfunction in up to 25% of patients, its pathogenesis remains unclear. Exposure of human iPSC-derived heart cells to SARS-CoV-2 revealed productive infection and robust transcriptomic and morphological signatures of damage, particularly in cardiomyocytes. -

Target Gene Gene Description Validation Diana Miranda
Supplemental Table S1. Mmu-miR-183-5p in silico predicted targets. TARGET GENE GENE DESCRIPTION VALIDATION DIANA MIRANDA MIRBRIDGE PICTAR PITA RNA22 TARGETSCAN TOTAL_HIT AP3M1 adaptor-related protein complex 3, mu 1 subunit V V V V V V V 7 BTG1 B-cell translocation gene 1, anti-proliferative V V V V V V V 7 CLCN3 chloride channel, voltage-sensitive 3 V V V V V V V 7 CTDSPL CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) small phosphatase-like V V V V V V V 7 DUSP10 dual specificity phosphatase 10 V V V V V V V 7 MAP3K4 mitogen-activated protein kinase kinase kinase 4 V V V V V V V 7 PDCD4 programmed cell death 4 (neoplastic transformation inhibitor) V V V V V V V 7 PPP2R5C protein phosphatase 2, regulatory subunit B', gamma V V V V V V V 7 PTPN4 protein tyrosine phosphatase, non-receptor type 4 (megakaryocyte) V V V V V V V 7 EZR ezrin V V V V V V 6 FOXO1 forkhead box O1 V V V V V V 6 ANKRD13C ankyrin repeat domain 13C V V V V V V 6 ARHGAP6 Rho GTPase activating protein 6 V V V V V V 6 BACH2 BTB and CNC homology 1, basic leucine zipper transcription factor 2 V V V V V V 6 BNIP3L BCL2/adenovirus E1B 19kDa interacting protein 3-like V V V V V V 6 BRMS1L breast cancer metastasis-suppressor 1-like V V V V V V 6 CDK5R1 cyclin-dependent kinase 5, regulatory subunit 1 (p35) V V V V V V 6 CTDSP1 CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) small phosphatase 1 V V V V V V 6 DCX doublecortin V V V V V V 6 ENAH enabled homolog (Drosophila) V V V V V V 6 EPHA4 EPH receptor A4 V V V V V V 6 FOXP1 forkhead box P1 V -
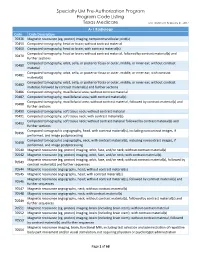
Specialty UM Pre-Authorization Program Code Listing
Specialty UM Pre-Authorization Program Program Code Listing Texas Medicare Last Updated: February 21, 2017 A-1 Radiology Code Code Description 70336 Magnetic resonance (eg, proton) imaging, temporomandibular joint(s) 70450 Computed tomography, head or brain; without contrast material 70460 Computed tomography, head or brain; with contrast material(s) Computed tomography, head or brain; without contrast material, followed by contrast material(s) and 70470 further sections Computed tomography, orbit, sella, or posterior fossa or outer, middle, or inner ear; without contrast 70480 material Computed tomography, orbit, sella, or posterior fossa or outer, middle, or inner ear; with contrast 70481 material(s) Computed tomography, orbit, sella, or posterior fossa or outer, middle, or inner ear; without contrast 70482 material, followed by contrast material(s) and further sections 70486 Computed tomography, maxillofacial area; without contrast material 70487 Computed tomography, maxillofacial area; with contrast material(s) Computed tomography, maxillofacial area; without contrast material, followed by contrast material(s) and 70488 further sections 70490 Computed tomography, soft tissue neck; without contrast material 70491 Computed tomography, soft tissue neck; with contrast material(s) Computed tomography, soft tissue neck; without contrast material followed by contrast material(s) and 70492 further sections Computed tomographic angiography, head, with contrast material(s), including noncontrast images, if 70496 performed, and image postprocessing -

Fncel-12-00217
ORIGINAL RESEARCH published: 03 August 2018 doi: 10.3389/fncel.2018.00217 ASD-Associated De Novo Mutations in Five Actin Regulators Show Both Shared and Distinct Defects in Dendritic Spines and Inhibitory Synapses in Cultured Hippocampal Neurons Iryna Hlushchenko 1, Pushpa Khanal 1, Amr Abouelezz 1,2,3, Ville O. Paavilainen 2,4 and Pirta Hotulainen 1* 1 Minerva Foundation Institute for Medical Research, Helsinki, Finland, 2 HiLIFE, University of Helsinki, Helsinki, Finland, 3 Neuroscience Center, University of Helsinki, Helsinki, Finland, 4 Institute of Biotechnology, University of Helsinki, Helsinki, Finland Edited by: Monica Mendes Sousa, Many actin cytoskeleton-regulating proteins control dendritic spine morphology and i3S, Instituto de Investigação e density, which are cellular features often altered in autism spectrum disorder (ASD). Inovação em Saúde, Portugal Recent studies using animal models show that autism-related behavior can be rescued Reviewed by: by either manipulating actin regulators or by reversing dendritic spine density or Marco Rust, Philipps University of Marburg, morphology. Based on these studies, the actin cytoskeleton is a potential target pathway Germany for developing new ASD treatments. Thus, it is important to understand how different Jaewon Ko, Daegu Gyeongbuk Institute of Science ASD-associated actin regulators contribute to the regulation of dendritic spines and and Technology (DGIST), South Korea how ASD-associated mutations modulate this regulation. For this study, we selected Maurizio Giustetto, five genes encoding different actin-regulating proteins and induced ASD-associated Università degli Studi di Torino, Italy de novo missense mutations in these proteins. We assessed the functionality of the *Correspondence: Pirta Hotulainen wild-type and mutated proteins by analyzing their subcellular localization, and by pirta.hotulainen@helsinki.fi analyzing the dendritic spine phenotypes induced by the expression of these proteins.