Genomics of Bacteria from an Ancient Marine Origin: Clues to Survival in an Oligotrophic Environment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Research Article Genomic Analysis of Bacillus Megaterium NCT-2 Reveals Its Genetic Basis for the Bioremediation of Secondary Salinization Soil
Hindawi International Journal of Genomics Volume 2020, Article ID 4109186, 11 pages https://doi.org/10.1155/2020/4109186 Research Article Genomic Analysis of Bacillus megaterium NCT-2 Reveals Its Genetic Basis for the Bioremediation of Secondary Salinization Soil Bin Wang ,1 Dan Zhang,1 Shaohua Chu ,1 Yuee Zhi,1 Xiaorui Liu ,2 and Pei Zhou 1 1School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai 200240, China 2The International Peace Maternity and Child Health Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China Correspondence should be addressed to Xiaorui Liu; [email protected] and Pei Zhou; [email protected] Received 11 November 2019; Revised 1 February 2020; Accepted 8 February 2020; Published 29 February 2020 Guest Editor: Ravi Kant Copyright © 2020 Bin Wang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Bacillus megaterium NCT-2 is a nitrate-uptake bacterial, which shows high bioremediation capacity in secondary salinization soil, including nitrate-reducing capacity, phosphate solubilization, and salinity adaptation. To gain insights into the bioremediation capacity at the genetic level, the complete genome sequence was obtained by using a multiplatform strategy involving HiSeq and PacBio sequencing. The NCT-2 genome consists of a circular chromosome of 5.19 Mbp and ten indigenous plasmids, totaling 5.88 Mbp with an average GC content of 37.87%. The chromosome encodes 5,606 genes, 142 tRNAs, and 53 rRNAs. Genes involved in the features of the bioremediation in secondary salinization soil and plant growth promotion were identified in the genome, such as nitrogen metabolism, phosphate uptake, the synthesis of organic acids and phosphatase for phosphate- solubilizing ability, and Trp-dependent IAA synthetic system. -

The Genome of Bacillus Coahuilensis Reveals Adaptations Essential for Survival in the Relic of an Ancient Marine Environment
The genome of Bacillus coahuilensis reveals adaptations essential for survival in the relic of an ancient marine environment Luis David Alcaraz*, Gabriela Olmedo*, Germa´ n Bonilla†, Rene´ Cerritos†, Gustavo Herna´ ndez‡, Alfredo Cruz‡, Enrique Ramı´rez§, Catherine Putonti¶, Beatriz Jime´nez*‡, Eva Martı´nez*,Varinia Lo´pez*, Jacqueline L. Arvizu*, Francisco Ayala*, Francisco Razo*, Juan Caballero*, Janet Siefertʈ, Luis Eguiarte†, Jean-Philippe Vielle*‡, Octavio Martı´nez*‡, Valeria Souza†, Alfredo Herrera-Estrella*‡, and Luis Herrera-Estrella*‡** ‡Laboratorio Nacional de Geno´mica para la Biodiversidad (Langebio), *Departamento de Ingenierı´aGene´tica and §Departamento de Biotecnologı´a, Cinvestav, Campus Guanajuato, AP 629 Irapuato, Guanajuato CP36500, Me´xico; †Departamento de Ecologı´aEvolutiva, Instituto de Ecologı´a,Universidad Nacional Auto´noma de Me´xico, CU, AP 70-275 Coyoaca´n 04510 Me´xico D.F., Me´xico; ʈDepartment of Statistics, Rice University, P.O. Box 1892, MS-138, Houston, TX 77251-1892; and ¶Departments of Computer Science and Biology and Biochemistry, 4800 Cullen Boulevard, University of Houston, Houston, TX 77204-5001 Contributed by Luis Herrera-Estrella, January 31, 2008 (sent for review December 10, 2007) The Cuatro Cie´negas Basin (CCB) in the central part of the Chihua- phylogenetic analysis of 16S rRNA sequences indicates that B. han desert (Coahuila, Mexico) hosts a wide diversity of microor- coahuilensis is closely related to other marine Bacillus spp. (4), in ganisms contained within springs thought to be geomorphological agreement with the theory of an ancient marine origin of these relics of an ancient sea. A major question remaining to be answered ponds. We sequenced the genome of B. -

Microevolution Analysis of Bacillus Coahuilensis Unveils Differences in Phosphorus Acquisition Strategies and Their Regulation
ORIGINAL RESEARCH published: 08 February 2016 doi: 10.3389/fmicb.2016.00058 Microevolution Analysis of Bacillus coahuilensis Unveils Differences in Phosphorus Acquisition Strategies and Their Regulation Zulema Gómez-Lunar1, Ismael Hernández-González1, María-Dolores Rodríguez-Torres1, Valeria Souza2 and Gabriela Olmedo-Álvarez1* 1 Laboratorio de Biología Molecular y Ecología Microbiana, Departamento de Ingeniería Genética, Unidad Irapuato, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Irapuato, Mexico, 2 Laboratorio de Evolución Molecular y Experimental, Departamento de Ecología Evolutiva, Instituto de Ecología, Universidad Nacional Autónoma de México, México City, Mexico Bacterial genomes undergo numerous events of gene losses and gains that generate genome variability among strains of the same species (microevolution). Our aim was Edited by: to compare the genomes and relevant phenotypes of three Bacillus coahuilensis Frank T. Robb, strains from two oligotrophic hydrological systems in the Cuatro Ciénegas Basin University of Maryland School (México), to unveil the environmental challenges that this species cope with, and the of Medicine, USA Reviewed by: microevolutionary differences in these genotypes. Since the strains were isolated from David John Studholme, a low P environment, we placed emphasis on the search of different phosphorus University of Exeter, UK acquisition strategies. The three B. coahuilensis strains exhibited similar numbers of Juan M. Gonzalez, Consejo Superior de Investigaciones coding DNA sequences, of which 82% (2,893) constituted the core genome, and 18% Científicas, Spain corresponded to accessory genes. Most of the genes in this last group were associated *Correspondence: with mobile genetic elements (MGEs) or were annotated as hypothetical proteins. Ten Gabriela Olmedo-Álvarez percent of the pangenome consisted of strain-specific genes. -

Centro De Investigación Y De Estudios Avanzados Del
CENTRO DE INVESTIGACIÓN Y DE ESTUDIOS AVANZADOS DEL INSTITUTO POLITÉCNICO NACIONAL UNIDAD IRAPUATO DEPARTAMENTO DE INGENIERÍA GENÉTICA AISLADOS DE COMUNIDADES DE SEDIMENTO DE CUATROCIÉNEGAS, COAHUILA COMO MODELO PARA ANALIZAR LA COHERENCIA FENOTÍPICA DEL GÉNERO BACILLUS Tesis que presenta M.C. MARIA DOLORES RODRIGUEZ TORRES Para Obtener el Grado de Doctora en Ciencias En la Especialidad de Biotecnología de Plantas Directora de la Tesis: Dra. Gabriela Olmedo Álvarez AGRADECIMIENTOS Agradezco el apoyo financiero otorgado por el CONACYT con la beca número 229493 para la realización de este trabajo de investigación. Al CINVESTAV por la oportunidad que me dieron para realizar mis estudios dentro de la institución y permitirme forjar la pasión por la investigación. A la Dra. Gabriela Olmedo Álvarez por su gran paciencia, su apoyo y permitirme trabajar en el laboratorio de Biología Molecular y Ecología Microbiana. A la Dra. Valeria Souza Saldívar por compartir su pasión por nuestro lugar de estudio: Cuatrociénegas, Coahuila. A los Doctores: Laila Pamela Partida Martínez, Jorge Eugenio Ibarra Rendón, Alexander de Luna Fors, Rafael Rivera Bustamante, por sus valiosas sugerencias y aportaciones a lo largo de estos años. A los miembros del Laboratorio de Metabolómica y Fisiología Molecular y al Laboratorio Biología de Sistemas Genéticos, por permitirme utilizar sus equipos. A la M.C. Africa Islas Robles por su dedicación en el trabajo, el apoyo brindado y la amistad. Al grupo de seminarios de microorganismos. A todos los colaboradores y amigos que participaron en la elaboración y diseño de experimentos, análisis y discusiones: Zulema Gómez Lunar, Ismael Hernández González, Varinia López, Julio Cruz Medina, José Antonio González, Adrian Jinich, Yunuen Tapia, Luis J. -

Bacillus Telluris Sp. Nov. Isolated from Greenhouse Soil in Beijing, China
microorganisms Article Bacillus telluris sp. nov. Isolated from Greenhouse Soil in Beijing, China He-Bao Guo, Shan-Wen He , Xing Wang, Kyu-Kyu Thin, Hai-Lei Wei * and Xiao-Xia Zhang * Key Laboratory of Microbial Resources Collection and Preservation, Ministry of Agriculture and Rural Affairs, Institute of Agricultural Resources and Regional Planning, Chinese Academy of Agricultural Sciences, Beijing 100081, China; [email protected] (H.-B.G.); [email protected] (S.-W.H.); [email protected] (X.W.); [email protected] (K.-K.H.) * Correspondence: [email protected] (H.-L.W.); [email protected] (X.-X.Z.) Received: 28 February 2020; Accepted: 8 May 2020; Published: 10 May 2020 Abstract: A novel Gram-stain-positive, rod-shaped, endospore-forming bacterium, which we designated as strain 03113T, was isolated from greenhouse soil in Beijing, China. Phylogenetic analysis based on 16S rRNA gene sequences showed strain 03113T is in the genus Bacillus and had T the highest similarity to Bacillus solani CCTCC AB 2014277 (98.14%). The strain grew at 4 ◦C–50 ◦C (optimum 37 ◦C), with 0–10% (w/v) NaCl (optimum 5%), and in the range of pH 3.0–12.0 (optimum pH 8.0). Menaquinone was identified as MK-7, and the major polar lipids were diphosphatidylglycerol, phosphatidylglycerol, and phosphatidylethanolamine. The main major cellular fatty acids detected were anteiso-C15:0 (51.35%) and iso-C15:0 (11.06%), which are the predominant cellular fatty acids found in all recognized members of the genus Bacillus. The 16S rRNA gene sequence and core-genome analysis, the average nucleotide identity (ANI), and in silico DNA—DNA hybridization (DDH) value between strain 03113T and the most closely related species were 70.5% and 22.6%, respectively, which supported our conclusion that 03113T represented a novel species in the genus Bacillus. -

The Pangenome Diversity, Dynamics and Evolution of Genomes the Pangenome Hervé Tettelin • Duccio Medini Editors
Hervé Tettelin Duccio Medini Editors The Pangenome Diversity, Dynamics and Evolution of Genomes The Pangenome Hervé Tettelin • Duccio Medini Editors The Pangenome Diversity, Dynamics and Evolution of Genomes Editors Hervé Tettelin Duccio Medini Department of Microbiology and GSK Vaccines R&D Immunology, Institute for Genome Siena, Italy Sciences University of Maryland School of Medicine Baltimore, Maryland, USA ISBN 978-3-030-38280-3 ISBN 978-3-030-38281-0 (eBook) https://doi.org/10.1007/978-3-030-38281-0 This book is an open access publication. © The Editor(s) (if applicable) and The Author(s) 2020. Open Access This book is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made. The images or other third party material in this book are included in the book’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the book’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -
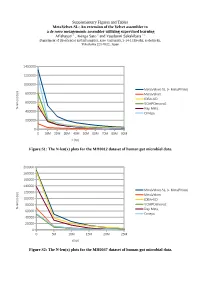
Supplementary Figures and Tables Metavelvet-SL
Supplementary Figures and Tables MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning Afiahayati 1 , Kengo Sato 1 and Yasubumi Sakakibara 1∗ Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan 1400000 1200000 1000000 MetaVelvet-SL (+ MetaPhlAn) ) p 800000 b MetaVelvet ( ) x ( IDBA-UD n 600000 e SOAPDenovo2 l - N Ray Meta 400000 Omega 200000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S1: The N-len(x) plots for the MH0012 dataset of human gut microbial data. 200000 180000 160000 140000 MetaVelvet-SL (+ MetaPhlAn) ) 120000 p b MetaVelvet ( ) 100000 x IDBA-UD ( n e l 80000 SOAPDenovo2 - N Ray Meta 60000 Omega 40000 20000 0 0 5M 10M 15M 20M 25M x(bp) Figure S2: The N-len(x) plots for the MH0047 dataset of human gut microbial data. 600000 500000 400000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 300000 x IDBA-UD ( n e l SOAPDenovo2 - N 200000 Ray Meta Omega 100000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S3: The N-len(x) plots for the SRS017227 dataset of human gut microbial data. 800000 700000 600000 500000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 400000 x IDBA-UD ( n e l SOAPDenovo2 - 300000 N Ray Meta 200000 Omega 100000 0 0 5M 10M 15M 20M 25M 30M 35M 40M 45M x (bp) Figure S4: The N-len(x) plots for the SRS018661 dataset of human gut microbial data. Formula to identify unique nodes in Velvet (Zerbino and Birney, 2008) ̄x2 ρ2− log2 2 F ( ̄x ,n,ρ )= +n 2 2 where ̄x = the coverage of node ρ = expected coverage of subgraph n = the length of node A node is “unique”, if its F > 5 . -
Variability of Rrna Operon Copy Number and Growth Rate Dynamics of Bacillus Isolated from an Extremely Oligotrophic Aquatic Ecosystem
ORIGINAL RESEARCH published: 05 January 2016 doi: 10.3389/fmicb.2015.01486 Variability of rRNA Operon Copy Number and Growth Rate Dynamics of Bacillus Isolated from an Extremely Oligotrophic Aquatic Ecosystem Jorge A. Valdivia-Anistro1, Luis E. Eguiarte-Fruns1, Gabriela Delgado-Sapién2, Pedro Márquez-Zacarías3, Jaime Gasca-Pineda1, Jennifer Learned4, James J. Elser4, Gabriela Olmedo-Alvarez5 and Valeria Souza1* 1 Laboratorio de Evolución Molecular y Experimental, Departamento de Ecología Evolutiva, Instituto de Ecología, Universidad Nacional Autónoma de México, Coyoacán, Mexico, 2 Laboratorio de Genómica Bacteriana, Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad Nacional Autónoma de México, Coyoacán, Mexico, 3 School of Biology, Georgia Institute of Technology, Atlanta, GA, USA, 4 School of Life Sciences, Arizona State University, Tempe, AZ, USA, 5 Laboratorio de Bacteriología Molecular, Departamento de Ingeniería Genética, CINVESTAV – Unidad Irapuato, Irapuato, Mexico The ribosomal RNA (rrn) operon is a key suite of genes related to the production Edited by: of protein synthesis machinery and thus to bacterial growth physiology. Experimental Roland Hatzenpichler, evidence has suggested an intrinsic relationship between the number of copies of this California Institute of Technology, USA operon and environmental resource availability, especially the availability of phosphorus Reviewed by: (P), because bacteria that live in oligotrophic ecosystems usually have few rrn operons Sebastian Kopf, Princeton University, USA and a slow growth rate. The Cuatro Ciénegas Basin (CCB) is a complex aquatic Albert Leopold Mueller, ecosystem that contains an unusually high microbial diversity that is able to persist Stanford University, USA under highly oligotrophic conditions. These environmental conditions impose a variety *Correspondence: Valeria Souza of strong selective pressures that shape the genome dynamics of their inhabitants. -

Bacillus Massiliosenegalensis Sp. Nov
Standards in Genomic Sciences (2013) 8:264-278 DOI:10.4056/sigs.3496989 Non contiguous-finished genome sequence and description of Bacillus massiliosenegalensis sp. nov. Dhamodharan Ramasamy1, Jean-Christophe Lagier1, Aurore Gorlas1, Didier Raoult1 and Pierre-Edouard Fournier1* 1Aix-Marseille Université, URMITE, Faculté de médecine, Marseille, France Corresponding author: Pierre-Edouard Fournier ([email protected]) Keywords: Bacillus massiliosenegalensis, genome, culturomics, taxono-genomics Bacillus massiliosenegalensis strain JC6T sp. nov. is the type strain of Bacillus massiliosenegalensis sp. nov., a new species within the genus Bacillus. This strain was isolat- ed from the fecal flora of a healthy Senegalese patient. B. massiliosenegalensis is an aerobic Gram-positive rod-shaped bacterium. Here we describe the features of this organism, togeth- er with the complete genome sequence and annotation. The 4,981,278-bp long genome comprises a 4,957,301-bp chromosome and a 23,977-bp plasmid. The chromosome contains 4,925 protein-coding and 72 RNA genes, including 4 rRNA genes. The plasmid contains 29 protein-coding genes. Introduction Bacillus massiliosenegalensis strain JC6T (= CSUR strain JC6T together with the description of the P151 = DSM 25957) is the type strain of B. complete genomic sequencing and annotation. massiliosenegalensis sp. nov., a new species within These characteristics support the creation of the the genus Bacillus. This bacterium is a Gram- species B. massiliosenegalensis. positive, aerobic, catalase-positive and indole- The genus Bacillus (Cohn 1872) was created in negative bacillus that was isolated from the stool 1872 [26] and currently consists of mainly Gram- of a healthy Senegalese patient as part of a study positive, motile, and spore-forming bacilli. -

Genome-Scale Evaluation of the Biotechnological Potential of Red Sea Bacilli Strains
Genome-scale Evaluation of the Biotechnological Potential of Red Sea Bacilli Strains Dissertation by Ghofran Othoum In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy King Abdullah University of Science and Technology Thuwal, Kingdom of Saudi Arabia February, 2018 2 EXAMINATION COMMITTEE PAGE The dissertation of Ghofran Othoum is approved by the examination committee. Committee Chairperson: Prof. Vladimir B. Bajic Committee Members: Prof. Alan Christoffels., Prof. Mani Sarathy, Prof. Takashi Gojobori 3 COPYRIGHT PAGE © February 2018 Ghofran Othoum All Rights Reserved 4 ABSTRACT Genome-scale Evaluation of the Biotechnological Potential of Red Sea Bacilli Strains Ghofran Othoum The increasing spectrum of multidrug-resistant bacteria has caused a major global public health concern, necessitating the discovery of novel antimicrobial agents. Additionally, recent advancements in the use of microbial cells for the scalable production of industrial enzymes has encouraged the screening of new environments for efficient microbial cell factories. The unique ecological niche of the Red Sea points to the promising metabolic and biosynthetic potential of its microbial system. Here, ten sequenced Bacilli strains, that are isolated from microbial mat and mangrove mud samples from the Red Sea, were evaluated for their use as platforms for protein production and biosynthesis of bioactive compounds. Two of the species (B. paralicheniformis Bac48 and B. litoralis Bac94) were found to secrete twice as much protein as Bacillus subtilis 168, and B. litoralis Bac94 had complete Tat and Sec protein secretion systems. Additionally, four Red Sea Species (B. paralicheniformis Bac48, Virgibacillus sp. Bac330, B. vallismortis Bac111, B. amyloliquefaciens Bac57) showed capabilities for genetic transformation and possessed competence genes. -
Effects of Food Changes on Intestinal Bacterial Diversity of Wintering Hooded Cranes (Grus Monacha)
animals Article Effects of Food Changes on Intestinal Bacterial Diversity of Wintering Hooded Cranes (Grus monacha) Nazhong Zhang 1,2 , Lizhi Zhou 1,2,* , Zhuqing Yang 1,2 and Jingjing Gu 1,2 1 School of Resources and Environmental Engineering, Anhui University, Hefei 230601, China; [email protected] (N.Z.); [email protected] (Z.Y.); [email protected] (J.G.) 2 Anhui Province Key Laboratory of Wetland Ecosystem Protection and Restoration, Anhui University, Hefei 230601, China * Correspondence: [email protected] Simple Summary: The intestinal microbiota plays a vital role in the health of animals, and food is an important factor that influences the intestinal microbial community. During the winter months, waterbirds require certain foods to supply them with energy through the cold winter. Due to changes in the plant resources available to waterbirds, their intestinal bacteria will vary accordingly. In this study, we analysed the relationship between food composition and intestinal bacteria in hooded cranes (Grus monacha). We found that food resources from similar habitats were more similar, and the corresponding hooded crane intestinal bacteria were also more similar. The results show that the intestinal bacteria of hooded cranes had a certain adaptability to the type of food being consumed. This study contributes novel insights into the diet of hooded cranes in the winter months, allowing for improved protection and management strategies. Abstract: As food is recognised as an important factor affecting the intestinal microbiota, seasonal changes in diet can influence the community composition. The hooded crane (Grus monacha) is an endangered migratory waterbird species, with some of the population wintering in the sallow lakes in Citation: Zhang, N.; Zhou, L.; Yang, the middle and lower Yangtze River floodplain. -

The Lost World of Cuatro Cie´ Negas Basin, a Relictual Bacterial Niche In
RESEARCH ARTICLE The lost world of Cuatro Cie´ negas Basin, a relictual bacterial niche in a desert oasis Valeria Souza1†*, Alejandra Moreno-Letelier2†, Michael Travisano3, Luis David Alcaraz4‡, Gabriela Olmedo5, Luis Enrique Eguiarte1* 1Departamento de Ecologı´a Evolutiva, Instituto de Ecologı´a, Universidad Nacional Auto´noma de Me´xico, Coyoaca´n, Mexico; 2Jardı´n Bota´nico, Instituto de Biologı´a Universidad Nacional Auto´noma de Me´xico, Coyoaca´n, Mexico; 3Department of Ecology, Evolution and Behavior, University of Minnesota, Saint Paul, United States; 4Laboratorio Nacional de la Ciencias de la Sostenibilidad, Instituto de Ecologı´a, Universidad Nacional Auto´noma de Me´xico, Coyoaca´n, Mexico; 5Laboratorio de Biologı´a Molecular y Ecologı´a Microbiana, Departamento de Ingenierı´a Gene´tica, Unidad Irapuato Centro de Investigacio´n y Estudios Avanzados, Guanajuato, Mexico Abstract Barriers to microbial migrations can lead adaptive radiations and increased endemism. We propose that extreme unbalanced nutrient stoichiometry of essential nutrients can be a barrier to microbial immigration over geological timescales. At the oasis in the Cuatro Cie´negas Basin in *For correspondence: Mexico, nutrient stoichiometric proportions are skewed given the low phosphorus availability in the [email protected] (VS); ecosystem. We show that this endangered oasis can be a model for a lost world. The ancient niche [email protected] (LEE) of extreme unbalanced nutrient stoichiometry favoured survival of ancestral microorganisms. This † These authors contributed extreme nutrient imbalance persisted due to environmental stability and low extinction rates, equally to this work generating a diverse and unique bacterial community. Several endemic clades of Bacillus invaded Present address: the Cuatro Cienegas region in two geological times, the late Precambrian and the Jurassic.