Investigating the Genetic Basis of Plumage Variation in the Australian Magpie, Cracticus Tibicen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biolphilately Vol-64 No-3
BIOPHILATELY OFFICIAL JOURNAL OF THE BIOLOGY UNIT OF ATA MARCH 2020 VOLUME 69, NUMBER 1 Great fleas have little fleas upon their backs to bite 'em, And little fleas have lesser fleas, and so ad infinitum. —Augustus De Morgan Dr. Indraneil Das Pangolins on Stamps More Inside >> IN THIS ISSUE NEW ISSUES: ARTICLES & ILLUSTRATIONS: From the Editor’s Desk ......................... 1 Botany – Christopher E. Dahle ............ 17 Pangolins on Stamps of the President’s Message .............................. 2 Fungi – Paul A. Mistretta .................... 28 World – Dr. Indraneil Das ..................7 Secretary -Treasurer’s Corner ................ 3 Mammalia – Michael Prince ................ 31 Squeaky Curtain – Frank Jacobs .......... 15 New Members ....................................... 3 Ornithology – Glenn G. Mertz ............. 35 New Plants in the Philatelic News of Note ......................................... 3 Ichthyology – J. Dale Shively .............. 57 Herbarium – Christopher Dahle ....... 23 Women’s Suffrage – Dawn Hamman .... 4 Entomology – Donald Wright, Jr. ........ 59 Rats! ..................................................... 34 Event Calendar ...................................... 6 Paleontology – Michael Kogan ........... 65 New Birds in the Philatelic Wedding Set ........................................ 16 Aviary – Charles E. Braun ............... 51 Glossary ............................................... 72 Biology Reference Websites ................ 69 ii Biophilately March 2020 Vol. 69 (1) BIOPHILATELY BIOLOGY UNIT -

The Avifauna of Mt. Karimui, Chimbu Province, Papua New Guinea, Including Evidence for Long-Term Population Dynamics in Undisturbed Tropical Forest
Ben Freeman & Alexandra M. Class Freeman 30 Bull. B.O.C. 2014 134(1) The avifauna of Mt. Karimui, Chimbu Province, Papua New Guinea, including evidence for long-term population dynamics in undisturbed tropical forest Ben Freeman & Alexandra M. Class Freeman Received 27 July 2013 Summary.—We conducted ornithological feld work on Mt. Karimui and in the surrounding lowlands in 2011–12, a site frst surveyed for birds by J. Diamond in 1965. We report range extensions, elevational records and notes on poorly known species observed during our work. We also present a list with elevational distributions for the 271 species recorded in the Karimui region. Finally, we detail possible changes in species abundance and distribution that have occurred between Diamond’s feld work and our own. Most prominently, we suggest that Bicolored Mouse-warbler Crateroscelis nigrorufa might recently have colonised Mt. Karimui’s north-western ridge, a rare example of distributional change in an avian population inhabiting intact tropical forests. The island of New Guinea harbours a diverse, largely endemic avifauna (Beehler et al. 1986). However, ornithological studies are hampered by difculties of access, safety and cost. Consequently, many of its endemic birds remain poorly known, and feld workers continue to describe new taxa (Prat 2000, Beehler et al. 2007), report large range extensions (Freeman et al. 2013) and elucidate natural history (Dumbacher et al. 1992). Of necessity, avifaunal studies are usually based on short-term feld work. As a result, population dynamics are poorly known and limited to comparisons of diferent surveys or diferences noticeable over short timescales (Diamond 1971, Mack & Wright 1996). -

Bear News/Make PDF & CD
International Bear News Quarterly Newsletter of the International Association for Bear Research and Management (IBA) and the IUCN/SSC Bear Specialist Group February 2002 vol. 11, no. 1 Contents Council News 2 From the President 4 USA/Russia Polar Bear Treaty Support 5 Secretary's Report 6 IBA Travel Grants Due 6 IBA Grants 6 IUCN Grants for Meetings 7 Frank Cooper Craighead, Jr. Eurasia 10 Curiosity Trapped the Cat! 10 Thailand Claws 11 Pyrenees Brown Bear Restoration 13 Genetic Analysis in Austria 13 Romania: Electric Fences & Livestock Americas 15 Peru Workshop & Grassroots Success 17 Andean Bears in Ecuador 18 Venezuela: Andean Bear Project 19 Andean Bear Website 20 Western USA 22 Southwest USA 24 Northcentral USA © Lance Craighead 25 Northeast USA In Memory: Frank Cooper Craighead, Jr. 25 Florida 26 Improved Black Bear Live Trap Publications Zoo News 36 Ursus 12 Contents 27 Polar Bear Management in Central Park 37 16th E. Black Bear Workshop Proceedings Student Affairs 37 Sloth Bears 28 Opportunity Notices 37 Ursus: COYS and Black Bears Banned! 28 Your Proposal is Your Friend Events Opportunities 38 Grizzly Bears: Small Populations Workshop 31 Bear Work Wanted 38 Eighth Western Black Bear Workshop 31 Bear Biologists Needed in Florida 38 Future IBA Conferences Education/Outreach 39 14th IBA Conf. Registration & Information 32 Call for Educational Materials IBA Bears in Culture 44 Contact Information 33 Kermode Bears: From Myth to Mutation 45 Membership Application & Survey 34 Bear Self-Medication 47 Publications Order Form IBA website: www.bearbiology.com Council News From the President Harry Reynolds For example, since 1993, the IBA findings of bear biology that make Alaska Department of Fish and Game grants program (formerly the Bevins science-based conservation an 1300 College Road grant review committee) has dis- effective tool in maintaining the Fairbanks, AK 99701, USA bursed US$108,000 on 30 projects to place of bears in our world. -

Dieter Thomas Tietze Editor How They Arise, Modify and Vanish
Fascinating Life Sciences Dieter Thomas Tietze Editor Bird Species How They Arise, Modify and Vanish Fascinating Life Sciences This interdisciplinary series brings together the most essential and captivating topics in the life sciences. They range from the plant sciences to zoology, from the microbiome to macrobiome, and from basic biology to biotechnology. The series not only highlights fascinating research; it also discusses major challenges associated with the life sciences and related disciplines and outlines future research directions. Individual volumes provide in-depth information, are richly illustrated with photographs, illustrations, and maps, and feature suggestions for further reading or glossaries where appropriate. Interested researchers in all areas of the life sciences, as well as biology enthusiasts, will find the series’ interdisciplinary focus and highly readable volumes especially appealing. More information about this series at http://www.springer.com/series/15408 Dieter Thomas Tietze Editor Bird Species How They Arise, Modify and Vanish Editor Dieter Thomas Tietze Natural History Museum Basel Basel, Switzerland ISSN 2509-6745 ISSN 2509-6753 (electronic) Fascinating Life Sciences ISBN 978-3-319-91688-0 ISBN 978-3-319-91689-7 (eBook) https://doi.org/10.1007/978-3-319-91689-7 Library of Congress Control Number: 2018948152 © The Editor(s) (if applicable) and The Author(s) 2018. This book is an open access publication. Open Access This book is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. -

Bird Abundances in Primary and Secondary Growths in Papua New Guinea: a Preliminary Assessment
Mongabay.com Open Access Journal - Tropical Conservation Science Vol.3 (4):373-388, 2010 Research Article Bird abundances in primary and secondary growths in Papua New Guinea: a preliminary assessment Kateřina Tvardíková1 1 Department of Zoology, Faculty of Biological Science, University of South Bohemia, Branišovská 31, CZ- 370 05 České Budějovice. Email: <[email protected] Abstract Papua New Guinea is the third largest remaining area of tropical forest after the Amazon and Congo basins. However, the growing intensity of large-scale slash-and-burn agriculture and logging call for conservation research to assess how local people´s traditional land-use practices result in conservation of local biodiversity, of which a species-rich and diverse component is the avian community. With this in mind, I conducted a preliminary survey of birds in small-scale secondary plots and in adjacent primary forest in Wanang Conservation Area in Papua New Guinea. I used mist-netting, point counts, and transect walks to compare the bird communities of 7-year-old secondary growth, and neighboring primary forest. The preliminary survey lasted 10 days and was conducted during the dry season (July) of 2008. I found no significant differences in summed bird abundances between forest types. However, species richness was higher in primary forest (98 species) than in secondary (78 species). The response of individual feeding guilds was also variable. Two habitats differed mainly in presence of canopy frugivores, which were more abundant (more than 80%) in primary than in secondary forests. A large difference (70%) was found also in understory and mid-story insectivores. Species occurring mainly in secondary forest were Hooded Butcherbird (Cracticus cassicus), Brown Oriole (Oriolus szalayi), and Helmeted Friarbird (Philemon buceroides). -

Australia's Biodiversity and Climate Change
Australia’s Biodiversity and Climate Change A strategic assessment of the vulnerability of Australia’s biodiversity to climate change A report to the Natural Resource Management Ministerial Council commissioned by the Australian Government. Prepared by the Biodiversity and Climate Change Expert Advisory Group: Will Steffen, Andrew A Burbidge, Lesley Hughes, Roger Kitching, David Lindenmayer, Warren Musgrave, Mark Stafford Smith and Patricia A Werner © Commonwealth of Australia 2009 ISBN 978-1-921298-67-7 Published in pre-publication form as a non-printable PDF at www.climatechange.gov.au by the Department of Climate Change. It will be published in hard copy by CSIRO publishing. For more information please email [email protected] This work is copyright. Apart from any use as permitted under the Copyright Act 1968, no part may be reproduced by any process without prior written permission from the Commonwealth. Requests and inquiries concerning reproduction and rights should be addressed to the: Commonwealth Copyright Administration Attorney-General's Department 3-5 National Circuit BARTON ACT 2600 Email: [email protected] Or online at: http://www.ag.gov.au Disclaimer The views and opinions expressed in this publication are those of the authors and do not necessarily reflect those of the Australian Government or the Minister for Climate Change and Water and the Minister for the Environment, Heritage and the Arts. Citation The book should be cited as: Steffen W, Burbidge AA, Hughes L, Kitching R, Lindenmayer D, Musgrave W, Stafford Smith M and Werner PA (2009) Australia’s biodiversity and climate change: a strategic assessment of the vulnerability of Australia’s biodiversity to climate change. -

Gymnorhina Tibicen Global Invasive
FULL ACCOUNT FOR: Gymnorhina tibicen Gymnorhina tibicen System: Terrestrial Kingdom Phylum Class Order Family Animalia Chordata Aves Passeriformes Cracticidae Common name Synonym Coracias tibicen Similar species Grallina cyanoleuca, Cracticus nigrogularis Summary Gymnorhina tibicen (the Australian magpie) was originally introduced to New Zealand around the 1860s in an attempt to combat pastoral pests. It is known to be extremely territorial, especially during the breeding season, and is known to assault other avian species and even humans. Magpies potentially threaten a number of indigenous avian fauna, as well as putting humans at risk of injury. view this species on IUCN Red List Species Description The Australian magpie (Gymnorhina tibicen), is a medium-sized ground-feeding passerine found throughout much of the Australian continent. They range from 36-44cm in length and weigh 280-340g with black and white plumage, black heads, wings and underparts together with white napes and shoulders (NRC, 1998). The iris of the adult magpie is red, whereas the juveniles' eyes are dark brown in colour. Minor differences exist between the male and female magpies, though in general, magpies are not considered to be sexually dimorphic (Simpson et al., 1993). Notes Although Australian magpies (Gymnorhina tibicen) have been known to have detrimental impacts on some birds, they can actually be beneficial to others. They do this by attacking and displacing common avian predators, such as harrier hawks and ravens, which in turn provides safe nesting grounds for a number of rural birds (Morgan et al, 2005). Lifecycle Stages The average life span of the Australian magpie (Gymnorhina tibicen) has not been studied in detail, but is estimated to be around 24 years, with some individuals living up to 30 years of age (Reilly, 1988). -

Paluma Nature Notes Contributions to the PDCA Newsletter, Turkey Talk, 2011-2018
Paluma Nature Notes Contributions to the PDCA Newsletter, Turkey Talk, 2011-2018 by Roy MacKay Compiled by Jamie Oliver Roy Mackay – Nature Notes Compilation Introduction Roy Mackay first moved to Paluma in 1987 having had a varied and successful career in Australia and PNG working as taxidermist, curator wildlife park director, photographer and book author. He was a highly skilled naturalist, with a keen interest in all plants and animals (especially in the tropics). Through his regular walks in and around Paluma and avid reading of the literature, Roy quickly became Paluma’s resident expert on the wildlife and natural history of the area. With the establishment of Paluma’s regular newsletter, Turkey Talk, Roy found an effective way to share this wealth of knowledge. His first contributions, starting with issue 28, dealt with short notes related to natural history (eg. announcements of new species lists, book reviews etc.) but by Issue 39, with an article entitled “Nature Notes” he concentrated on recording new and interesting observations from Paluma and nearby areas. With the exception of a series of 4 articles titled “Roy's Rovings” (under a different Turkey Talk editor), and the occasional missed article due to absence or ill health, and one article named “Paluma in the Clouds” Roy’s contributions were simply labelled “Nature Notes” and they became a much-read part of virtually all issues of Turkey Talk from number 45 onwards. In total he wrote 78 articles. All of these are reproduced below. Jamie Oliver December, 2019 Page 2 Contents Each Nature Notes article has been extracted from an issue of the newsletter of the Paluma & District Community Association (Turkey Talk – abbreviated at TT throughout). -

Ultimate Papua New Guinea Ii
The fantastic Forest Bittern showed memorably well at Varirata during this tour! (JM) ULTIMATE PAPUA NEW GUINEA II 25 AUGUST – 11 / 15 SEPTEMBER 2019 LEADER: JULIEN MAZENAUER Our second Ultimate Papua New Guinea tour in 2019, including New Britain, was an immense success and provided us with fantastic sightings throughout. A total of 19 Birds-of-paradise (BoPs), one of the most striking and extraordinairy bird families in the world, were seen. The most amazing one must have been the male Blue BoP, admired through the scope near Kumul lodge. A few females were seen previously at Rondon Ridge, but this male was just too much. Several males King-of-Saxony BoP – seen displaying – ranked high in our most memorable moments of the tour, especially walk-away views of a male obtained at Rondon Ridge. Along the Ketu River, we were able to observe the full display and mating of another cosmis species, Twelve-wired BoP. Despite the closing of Ambua, we obtained good views of a calling male Black Sicklebill, sighted along a new road close to Tabubil. Brown Sicklebill males were seen even better and for as long as we wanted, uttering their machine-gun like calls through the forest. The adult male Stephanie’s Astrapia at Rondon Ridge will never be forgotten, showing his incredible glossy green head colours. At Kumul, Ribbon-tailed Astrapia, one of the most striking BoP, amazed us down to a few meters thanks to a feeder especially created for birdwatchers. Additionally, great views of the small and incredible King BoP delighted us near Kiunga, as well as males Magnificent BoPs below Kumul. -

Hollis Taylor: Violinist/Composer/Zoömusicologist
Hollis Taylor: violinist/composer/zoömusicologist Hollis Taylor conducting fieldwork: Newhaven Station, Central Australia, which adjoins Aboriginal freehold land on all sides. (Photo: Jon Rose) Major research: the pied butcherbird (Cracticus nigrogularis). (see “Zoömusicologists” page for audio link Pied butcherbird.mp3) A pied butcherbird, Burleigh Heads, Queensland. (Photo: Hollis Taylor) Other research interests: lyrebirds, bowerbirds, Australian magpies, grey butcherbirds, currawongs, & animal aesthetics. Left: The interior decorations of a western bowerbird’s avenue bower (Alice Springs). His color preferences are green, white, and shiny objects. Right: The exterior decorations of a great bowerbird’s avenue bower (Darwin) display his similar color preferences. (Photos: Hollis Taylor) Satin bowerbirds prefer things blue, with a secondary preference to the color yellow (Blue Mountains, New South Wales). (Photo: Hollis Taylor) Hollis writes: Bowerbird males are vocalists, dancers, architects, interior decorators, collectors, landscape architects, and painters. And bowerbird females are art critics, contemplating what the other sex has created. Selected Publications Taylor, Hollis. 2011. The Australian Pied Butcherbird: The Composer's Muse. Music Forum: Journal of the Music Council of Australia 17 (2). Taylor, Hollis. 2011. Composers’ appropriation of pied butcherbird song: Henry Tate’s ‘undersong of Australia’ comes of age. Journal of Music Research Online. Taylor, Hollis. 2010. Blowin' in Birdland: Improvisation and the Australian pied butcherbird. Leonardo Music Journal 20: 79-83. Taylor, Hollis. 2009. Super Tweeter. Art Monthly Australia 225: 16-19. Taylor, Hollis. 2009. Olivier Messiaen's transcription of the Albert's lyrebird. AudioWings 12 (1): 2-5. Taylor, Hollis. 2009. Location, location, location: Auditioning the vocalizations of the Australian pied butcherbird. Soundscape: The Journal of Acoustic Ecology 9 (1): 14-16. -

Spirit Bears Canada Travel Brochure with Itinerary and Photos
Land of the Spirit Bears September 14 – 26, 2015 Cocver: Ignacio Yúfera Expedition Overview Known as Canada’s “forgotten coast,” British Columbia’s Great Bear Rainforest evokes a bygone time, with its ancient firs and Sitka Spruce towering 350 feet into the air, providing cover for its unique and mythical-seeming animal subspecies, like the white Kermode, or Spirit, Bear. All of the wildlife present when Captain Vancouver sailed here in 1793, still exists, and in abundance. Virtually roadless, it’s a land of densely forested islands and mist-shrouded fjords that reach back into the glacier-capped Coastal Mountain range. Our expedition will take you along BC’s central coast in search of the elusive Ker- © Tom Rivest © Tom mode, as well as Grizzly and black bears, wolves, eagles, and Hump- Photos: (Cover) Kermode, or Spirit, back Whales. Soak up the culture of the Tshimshian and Heiltsuk Bear, Island Odyssey, Grizzly Bears. First Nations people, who are working diligently to protect this, the largest remaining untouched coastal rainforest on the Pacific Coast. WWW.APEX- EXPEDITIONS.COM 800.861.6425 / 206.669.9272 © Ignacio Yúfera Itinerary Monday, September 14: Vancouver / Bella Bella Fly this morning from Vancouver to Bella Bella, a remote island village in central British Columbia, the traditional home of the Heiltsuk First Nation and the “Gateway to the Great Bear Rainforest”. A shuttle will meet you at the airport and take you to the marina where you board the water taxi for the Shearwater Resort, located on its own island just west of Bella Bella. Settle in for a welcome dinner and overnight. -
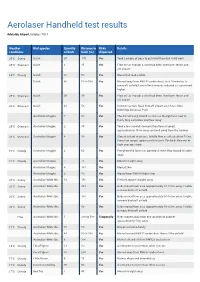
Aerolaser Handheld Test Results
Aerolaser Handheld test results Adelaide Airport October 2014 Weather Bird species Quantity Distance to Birds Details conditions of birds birds (m) dispersed 20°C Sunny Galah 20 170 Yes Took a couple of goes to get rid of them but it did work 20°C Overcast Galah 4 35 Yes Flew off as though a shot had been fired over fence and off airport 14°C Cloudy Galah 30 60 Yes Moved but took a while Galah 30 50 to 500 Yes Moved away from RWY12 undershoot, took 10 minutes to move off airfield, laser effectivenesds reduced as sun moved higher 20°C Overcast Galah 20 30 Yes Flew off as though a shot had been fired over fence and off airport 20°C Overcast Galah 40 50 Yes Instant reaction, flock flew off airport over fence intro Burbridge Business Park Australian Magpie 7 50 Yes The did not hang around as soon as the light was next to them, they carried on and flew away 20°C Overcast Australian Magpie 2 30 Yes Took a few seconds to react then flew at speed approximately 150m away and well away from the runway 20°C Overcast Australian Magpie 8 30 Yes Almost instant reactions. Initally flew as a flock about 100m. From that range I again used the laser. The birds flew out of sight over ops store 15°C Cloudy Australian Magpie 4 60 Yes Everytime the laser was pointed at them they moved straight away 15°C Cloudy Australian Magpie 7 70 Yes Moved straight away Australian Magpie 8 100 Yes Moved 24m Australian Magpie 2 50 Yes Moved from RWY05 flight strip 20°C Sunny Australian White Ibis 13 150 Yes Fled off airport straight away 20°C Sunny Australian White Ibis 250 Yes Birds moved from area approximately 50-100m away.