Integrated Genomics and Therapeutics Predictors of Small
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table of Contents (PDF)
July 26, 2011 u vol. 108 u no. 30 u 12187–12560 Cover image: Pictured is a Tasmanian devil (Sarcophilus harrisii), a carnivorous marsupial whose numbers are dwindling due to an infectious facial cancer called Devil Facial Tumor Disease. Webb Miller et al. sequenced the genome of devils from northwest and south- east Tasmania, spanning the range of this threatened species on the Australian island. The authors report that the sequences reveal a worrisome dearth of genetic diversity among devils, suggesting the need for genetically characterized stocks to help breed hardier devils that might be better equipped to fight diseases. See the article by Miller et al. on pages 12348–12353. Image courtesy of Stephan C. Schuster. From the Cover 12348 Decoding the Tasmanian devil genome 12283 Illuminating chromosomal architecture 12295 Symmetry of cultured cells 12319 Caloric restriction and infertility 12366 Genetic diversity among ants Contents COMMENTARIES 12189 Methyl fingerprinting of the nucleosome reveals the molecular mechanism of high-mobility group THIS WEEK IN PNAS nucleosomal-2 (HMGN2) association Catherine A. Musselman and Tatiana G. Kutateladze See companion article on page 12283 12187 In This Issue 12191 Examining the establishment of cellular axes using intrinsic chirality LETTERS (ONLINE ONLY) Jason C. McSheene and Rebecca D. Burdine See companion article on page 12295 E341 Difference between restoring and predicting 3D 12193 Secrets of palm oil biosynthesis revealed structures of the loops in G-protein–coupled Toni Voelker receptors by molecular modeling See companion article on page 12527 Gregory V. Nikiforovich, Christina M. Taylor, Garland R. Marshall, and Thomas J. Baranski E342 Reply to Nikiforovich et al.: Restoration of the loop regions of G-protein–coupled receptors Dahlia A. -

Table S1 the Four Gene Sets Derived from Gene Expression Profiles of Escs and Differentiated Cells
Table S1 The four gene sets derived from gene expression profiles of ESCs and differentiated cells Uniform High Uniform Low ES Up ES Down EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol 269261 Rpl12 11354 Abpa 68239 Krt42 15132 Hbb-bh1 67891 Rpl4 11537 Cfd 26380 Esrrb 15126 Hba-x 55949 Eef1b2 11698 Ambn 73703 Dppa2 15111 Hand2 18148 Npm1 11730 Ang3 67374 Jam2 65255 Asb4 67427 Rps20 11731 Ang2 22702 Zfp42 17292 Mesp1 15481 Hspa8 11807 Apoa2 58865 Tdh 19737 Rgs5 100041686 LOC100041686 11814 Apoc3 26388 Ifi202b 225518 Prdm6 11983 Atpif1 11945 Atp4b 11614 Nr0b1 20378 Frzb 19241 Tmsb4x 12007 Azgp1 76815 Calcoco2 12767 Cxcr4 20116 Rps8 12044 Bcl2a1a 219132 D14Ertd668e 103889 Hoxb2 20103 Rps5 12047 Bcl2a1d 381411 Gm1967 17701 Msx1 14694 Gnb2l1 12049 Bcl2l10 20899 Stra8 23796 Aplnr 19941 Rpl26 12096 Bglap1 78625 1700061G19Rik 12627 Cfc1 12070 Ngfrap1 12097 Bglap2 21816 Tgm1 12622 Cer1 19989 Rpl7 12267 C3ar1 67405 Nts 21385 Tbx2 19896 Rpl10a 12279 C9 435337 EG435337 56720 Tdo2 20044 Rps14 12391 Cav3 545913 Zscan4d 16869 Lhx1 19175 Psmb6 12409 Cbr2 244448 Triml1 22253 Unc5c 22627 Ywhae 12477 Ctla4 69134 2200001I15Rik 14174 Fgf3 19951 Rpl32 12523 Cd84 66065 Hsd17b14 16542 Kdr 66152 1110020P15Rik 12524 Cd86 81879 Tcfcp2l1 15122 Hba-a1 66489 Rpl35 12640 Cga 17907 Mylpf 15414 Hoxb6 15519 Hsp90aa1 12642 Ch25h 26424 Nr5a2 210530 Leprel1 66483 Rpl36al 12655 Chi3l3 83560 Tex14 12338 Capn6 27370 Rps26 12796 Camp 17450 Morc1 20671 Sox17 66576 Uqcrh 12869 Cox8b 79455 Pdcl2 20613 Snai1 22154 Tubb5 12959 Cryba4 231821 Centa1 17897 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Using Cellminer 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60 William C. Reinhold1, Margot Sunshine1,2, Sudhir
Author Manuscript Published OnlineFirst on June 5, 2015; DOI: 10.1158/1078-0432.CCR-15-0335 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60 William C. Reinhold1, Margot Sunshine1,2, Sudhir Varma1,2,3, James H. Doroshow1,4, and Yves Pommier1 1Developmental Therapeutics Branch and Laboratory of Molecular Pharmacology, Center for Cancer Research, NCI, NIH, Bethesda, Maryland; 2Systems Research and Applications Corp., Fairfax, Virginia; 3HiThru Analytics LLC, Laurel, Maryland; 4Developmental Therapeutics Program, DCTD, NCI, NIH, Bethesda, Maryland. Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). W.C. Reinhold and Y. Pommier share senior authorship. Corresponding Authors: William C. Reinhold, NIH, 9000 Rockville Pike, Building 37, Room 5041, Bethesda, MD 20892; E-mail: [email protected], and Yves Pommier, NIH, 9000 Rockville Pike, Building 37, Room 5068, Bethesda, MD 20892; E-mail [email protected] Running Title: CellMiner for Systems Pharmacology Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed. Downloaded from clincancerres.aacrjournals.org on September 27, 2021. © 2015 American Association for Cancer Research. Author Manuscript Published OnlineFirst on June 5, 2015; DOI: 10.1158/1078-0432.CCR-15-0335 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract The NCI-60 cancer cell line panel provides a premier model for data integration and systems pharmacology being the largest publicly available database of anticancer drug activity, , genomic, molecular, and phenotypic data. -

2016 Joint Meeting Program
April 15 – 17, 2016 Fairmont Chicago Millennium Park • Chicago, Illinois The AAP/ASCI/APSA conference is jointly provided by Boston University School of Medicine and AAP/ASCI/APSA. Meeting Program and Abstracts www.jointmeeting.org www.jointmeeting.org Special Events at the 2016 AAP/ASCI/APSA Joint Meeting Friday, April 15 Saturday, April 16 ASCI President’s Reception ASCI Food and Science Evening 6:15 – 7:15 p.m. 6:30 – 9:00 p.m. Gold Room The Mid-America Club, Aon Center ASCI Dinner & New Member AAP Member Banquet Induction Ceremony (Ticketed guests only) (Ticketed guests only) 7:00 – 10:00 p.m. 7:30 – 9:45 p.m. Imperial Ballroom, Level B2 Rouge, Lobby Level How to Solve a Scientific Puzzle: Speaker: Clara D. Bloomfield, MD Clues from Stockholm and Broadway The Ohio State University Comprehensive Cancer Center Speaker: Joe Goldstein, MD APSA Welcome Reception & University of Texas Southwestern Medical Center at Dallas Presidential Address APSA Dinner (Ticketed guests only) 9:00 p.m. – Midnight Signature Room, 360 Chicago, 7:30 – 9:00 p.m. John Hancock Center (off-site) Rouge, Lobby Level Speaker: Daniel DelloStritto, APSA President Finding One’s Scientific Niche: Musings from a Clinical Neuroscientist Speaker: Helen Mayberg, MD, Emory University Dessert Reception (open to all attendees) 10:00 p.m. – Midnight Imperial Foyer, Level B2 Sunday, April 17 APSA Future of Medicine and www.jointmeeting.org Residency Luncheon Noon – 2:00 p.m. Rouge, Lobby Level 2 www.jointmeeting.org Program Contents General Program Information 4 Continuing Medical Education Information 5 Faculty and Speaker Disclosures 7 Scientific Program Schedule 9 Speaker Biographies 16 Call for Nominations: 2017 Harrington Prize for Innovation in Medicine 26 AAP/ASCI/APSA Joint Meeting Faculty 27 Award Recipients 29 Call for Nominations: 2017 Harrington Scholar-Innovator Award 31 Call for Nominations: George M. -

Genome-Wide Association Analysis Reveal the Genetic Reasons Affect Melanin Spot Accumulation in Beak Skin of Ducks
Genome-wide association analysis reveal the genetic reasons affect melanin spot accumulation in beak skin of ducks Hehe Liu Sichuan Agricultural University Jianmei Wang Sichuan Agricultural University Jian Hu Chinese Academy of Agricultural Sciences Lei Wang Sichuan Agricultural University Zhanbao Guo Chinese Academy of Agricultural Sciences Wenlei Fan Chinese Academy of Agricultural Sciences Yaxi Xu Chinese Academy of Agricultural Sciences Dapeng Liu Chinese Academy of Agricultural Sciences Yunsheng Zhang Chinese Academy of Agricultural Sciences Ming Xie Chinese Academy of Agricultural Sciences Jing Tang Chinese Academy of Agricultural Sciences Wei Huang Chinese Academy of Agricultural Sciences Qi Zhang Chinese Academy of Agricultural Sciences Zhengkui Zhou Chinese Academy of Agricultural Sciences Shuisheng Hou ( [email protected] ) Chinese Academy of Agricultural Sciences Page 1/21 Research Article Keywords: skin melanin spot, duck, GWAS, genetic Posted Date: June 25th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-608516/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/21 Abstract Background Skin pigmentation is a broadly appearing phenomenon of most animals and humans in nature. Here we used a bird model to investigate why melanin spot deposits on the skin. Results We result shown that melanin deposition in bird skin was induced by growth age and ultraviolet UV radiation and determined by genetic factors. GWAS helped us to identify two major loci affecting melanin deposition, located on chromosomes 13 and 25, respectively. Fine mapping works narrowed the candidate regions to 0.98 Mb and 1.0 Mb on chromosome 13 and 25, respectively. -

SLFN11 Promotes CDT1 Degradation by CUL4 in Response to Replicative DNA Damage, While Its Absence Leads to Synthetic Lethality with ATR/CHK1 Inhibitors
SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors Ukhyun Joa,1, Yasuhisa Muraia, Sirisha Chakkab, Lu Chenb, Ken Chengb, Junko Muraic, Liton Kumar Sahaa, Lisa M. Miller Jenkinsd, and Yves Pommiera,1 aDevelopmental Therapeutics Branch, Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20814; bNational Center for Advancing Translational Sciences, Functional Genomics Laboratory, NIH, Rockville, MD 20850; cInstitute for Advanced Biosciences, Keio University, 997-0052 Yamagata, Japan; and dLaboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD 20892 Edited by Richard D. Kolodner, Ludwig Institute for Cancer Research, La Jolla, CA, and approved December 8, 2020 (received for review July 29, 2020) Schlafen-11 (SLFN11) inactivation in ∼50% of cancer cells confers condensation related to deposition of H3K27me3 in the gene broad chemoresistance. To identify therapeutic targets and under- body of SLFN11 by EZH2, a histone methyltransferase (11). lying molecular mechanisms for overcoming chemoresistance, we Targeting epigenetic regulators is therefore an attractive com- performed an unbiased genome-wide RNAi screen in SLFN11-WT bination strategy to overcome chemoresistance of SLFN11- and -knockout (KO) cells. We found that inactivation of Ataxia deficient cancers (10, 25, 26). An alternative approach is to at- Telangiectasia- and Rad3-related (ATR), CHK1, BRCA2, and RPA1 tack SLFN11-negative cancer cells by targeting the essential SLFN11 overcome chemoresistance to camptothecin (CPT) in -KO pathways that cells use to overcome replicative damage and cells. Accordingly, we validate that clinical inhibitors of ATR replication stress. -

POU Domain Family Values: Flexibility, Partnerships, and Developmental Codes
Downloaded from genesdev.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW POU domain family values: flexibility, partnerships, and developmental codes Aimee K. Ryan and Michael G. Rosenfeld 1 Howard Hughes Medical Institute, Department and School of Medicine, University of California at San Diego, La Jolla, Califiornia 92093-0648 USA Transcription factors serve critical roles in the progres- primary focus of this review. Crystallization of the Oct-1 sive development of general body plan, organ commit- and Pit-1 POU domains on DNA and in vitro studies ment, and finally, specific cell types. It has been the hope examining the specificity of POU domain cofactor inter- of most developmental biologists that comparison of the actions suggest that the flexibility with which the POU biological roles of a series of individual members within domain recognizes DNA-binding sites is a critical com- a family will permit at least some predictive generaliza- ponent of its ability to regulate gene expression. The tions regarding the developmental events that are likely implications of these data with respect to the mecha- to be regulated by a particular class of transcription fac- nisms utilized by the POU domain family of transcrip- tors. Here, we present an overview of the developmental tion factors to control developmental events will also be functions of the family of transcription factors charac- discussed. terized by the POU DNA-binding motif afforded by re- cent in vivo studies. Conformation of the POU domain on DNA The POU domain family of transcription factors was defined following the observation that the products of High-affinity site-specific DNA binding by POU domain three mammalian genes, Pit-l, Oct-l, and Oct-2 and the transcription factors requires both the POU-specific do- protein encoded by the Caenorhabditis elegans gene main and the POU homeodomain (Sturm and Herr 1988; unc-86 shared a region of homology, known as the POU Ingraham et al. -

Supplementary Data
SUPPLEMENTARY DATA A cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma Santiago Demajo et al. 1 SUPPLEMENTARY DATA INDEX Supplementary Methods p. 3 Supplementary References p. 8 Supplementary Tables (S1 to S5) p. 9 Supplementary Figures (S1 to S15) p. 17 2 SUPPLEMENTARY METHODS Western blot, immunoprecipitation, and qRT-PCR Western blot (WB) analysis was performed as previously described (1), using cyclin D1 (Santa Cruz Biotechnology, sc-753, RRID:AB_2070433) and tubulin (Sigma-Aldrich, T5168, RRID:AB_477579) antibodies. Co-immunoprecipitation assays were performed as described before (2), using cyclin D1 antibody (Santa Cruz Biotechnology, sc-8396, RRID:AB_627344) or control IgG (Santa Cruz Biotechnology, sc-2025, RRID:AB_737182) followed by protein G- magnetic beads (Invitrogen) incubation and elution with Glycine 100mM pH=2.5. Co-IP experiments were performed within five weeks after cell thawing. Cyclin D1 (Santa Cruz Biotechnology, sc-753), E2F4 (Bethyl, A302-134A, RRID:AB_1720353), FOXM1 (Santa Cruz Biotechnology, sc-502, RRID:AB_631523), and CBP (Santa Cruz Biotechnology, sc-7300, RRID:AB_626817) antibodies were used for WB detection. In figure 1A and supplementary figure S2A, the same blot was probed with cyclin D1 and tubulin antibodies by cutting the membrane. In figure 2H, cyclin D1 and CBP blots correspond to the same membrane while E2F4 and FOXM1 blots correspond to an independent membrane. Image acquisition was performed with ImageQuant LAS 4000 mini (GE Healthcare). Image processing and quantification were performed with Multi Gauge software (Fujifilm). For qRT-PCR analysis, cDNA was generated from 1 µg RNA with qScript cDNA Synthesis kit (Quantabio). qRT–PCR reaction was performed using SYBR green (Roche). -

HER2 Gene Signatures: (I) Novel and (Ii) Established by Desmedt Et Al 2008 (31)
Table S1: HER2 gene signatures: (i) Novel and (ii) Established by Desmedt et al 2008 (31). Pearson R [neratinib] is the correlation with neratinib response using a pharmacogenomic model of breast cancer cell lines (accessed online via CellMinerCDB). *indicates significantly correlated genes. n/a = data not available in CellMinerCDB Genesig N Gene ID Pearson R [neratinib] p-value (i) Novel 20 ERBB2 0.77 1.90E-08* SPDEF 0.45 4.20E-03* TFAP2B 0.2 0.24 CD24 0.38 0.019* SERHL2 0.41 0.0097* CNTNAP2 0.12 0.47 RPL19 0.29 0.073 CAPN13 0.51 1.00E-03* RPL23 0.22 0.18 LRRC26 n/a n/a PRODH 0.42 9.00E-03* GPRC5C 0.44 0.0056* GGCT 0.38 1.90E-02* CLCA2 0.31 5.70E-02 KDM5B 0.33 4.20E-02* SPP1 -0.25 1.30E-01 PHLDA1 -0.54 5.30E-04* C15orf48 0.06 7.10E-01 SUSD3 -0.09 5.90E-01 SERPINA1 0.14 4.10E-01 (ii) Established 24 ERBB2 0.77 1.90E-08* PERLD1 0.77 1.20E-08* PSMD3 0.33 0.04* PNMT 0.33 4.20E-02* GSDML 0.4 1.40E-02* CASC3 0.26 0.11 LASP1 0.32 0.049* WIPF2 0.27 9.70E-02 EPN3 0.42 8.50E-03* PHB 0.38 0.019* CLCA2 0.31 5.70E-02 ORMDL2 0.06 0.74 RAP1GAP 0.53 0.00059* CUEDC1 0.09 0.61 HOXC11 0.2 0.23 CYP2J2 0.45 0.0044* HGD 0.14 0.39 ABCA12 0.07 0.67 ATP2C2 0.42 0.0096* ITGA3 0 0.98 CEACAM5 0.4 0.012* TMEM16K 0.15 0.37 NR1D1 n/a n/a SNX7 -0.28 0.092 FJX1 -0.26 0.12 KCTD9 -0.11 0.53 PCTK3 -0.04 0.83 CREG1 0.17 0.3 Table S2: Up-regulated genes from the top 500 DEGs for each comparison by WAD score METABRIC METABRIC METABRIC TCGA ERBB2amp ERBB2mut oncERBB2mut HER2+ ERBB2 PIP ANKRD30A ERBB2 GRB7 CYP4Z1 CYP4Z1 SCGB2A2 PGAP3 PROM1 LRRC26 SPDEF GSDMB CD24 PPP1R1B FOXA1 -
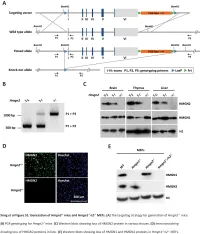
Supplemental Data.Pdf
Table S1. Summary of sequencing results A. DNase‐seq data % Align % Mismatch % >=Q30 bases Sample ID # Reads % unique reads (PF) Rate (PF) (PF) WT_1 84,301,522 86.76 0.52 93.34 96.30 WT_2 98,744,222 84.97 0.51 93.75 89.94 Hmgn1‐/‐_1 79,620,656 83.94 0.86 90.58 88.65 Hmgn1‐/‐_2 62,673,782 84.13 0.87 91.30 89.18 Hmgn2‐/‐_1 87,734,440 83.49 0.71 91.81 90.00 Hmgn2‐/‐_2 82,498,808 83.25 0.69 92.73 90.66 Hmgn1‐/‐n2‐/‐_1 71,739,638 68.51 2.31 81.11 89.22 Hmgn1‐/‐n2‐/‐_2 74,113,682 68.19 2.37 81.16 86.57 B. ChIP‐seq data Histone % Align % Mismatch % >=Q30 % unique Genotypes # Reads marks (PF) Rate (PF) bases (PF) reads H3K4me1 100670054 92.99 0.28 91.21 87.29 H3K4me3 67064272 91.97 0.35 89.11 27.15 WT H3K27ac 90,340,242 93.57 0.28 95.02 89.80 input 111,292,572 78.24 0.55 96.07 86.99 H3K4me1 84598176 92.34 0.33 91.2 81.69 H3K4me3 90032064 92.19 0.44 88.76 15.81 Hmgn1‐/‐n2‐/‐ H3K27ac 86,260,526 93.40 0.29 94.94 87.49 input 78,142,334 78.47 0.56 95.82 81.11 C. MNase‐seq data % Mismatch % >=Q30 bases % unique Sample ID # Reads % Align (PF) Rate (PF) (PF) reads WT_1_Extensive 45,232,694 55.23 1.49 90.22 81.73 WT_1_Limited 105,460,950 58.03 1.39 90.81 79.62 WT_2_Extensive 40,785,338 67.34 1.06 89.76 89.60 WT_2_Limited 105,738,078 68.34 1.05 90.29 85.96 Hmgn1‐/‐n2‐/‐_1_Extensive 117,927,050 55.74 1.49 89.50 78.01 Hmgn1‐/‐n2‐/‐_1_Limited 61,846,742 63.76 1.22 90.57 84.55 Hmgn1‐/‐n2‐/‐_2_Extensive 137,673,830 60.04 1.30 89.28 78.99 Hmgn1‐/‐n2‐/‐_2_Limited 45,696,614 62.70 1.21 90.71 85.52 D. -

POU2F3 (NM 014352) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC210969L1 POU2F3 (NM_014352) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: POU2F3 (NM_014352) Human Tagged ORF Clone Tag: Myc-DDK Symbol: POU2F3 Synonyms: Epoc-1; OCT-11; OCT11; OTF-11; PLA-1; PLA1; Skn-1a Vector: pLenti-C-Myc-DDK (PS100064) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: None ORF Nucleotide The ORF insert of this clone is exactly the same as(RC210969). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_014352 ORF Size: 1308 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 POU2F3 (NM_014352) Human Tagged ORF Clone – RC210969L1 OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_014352.1 RefSeq Size: 3013 bp RefSeq ORF: 1311 bp Locus ID: 25833 UniProt ID: Q9UKI9 MW: 47.4 kDa Gene Summary: This gene encodes a member of the POU domain family of transcription factors.