Electroorganic Synthesis for the Conversion of Fatty Acids and Levulinic Acid Into Chemicals and Alternative Fuels
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Retention Indices for Frequently Reported Compounds of Plant Essential Oils
Retention Indices for Frequently Reported Compounds of Plant Essential Oils V. I. Babushok,a) P. J. Linstrom, and I. G. Zenkevichb) National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA (Received 1 August 2011; accepted 27 September 2011; published online 29 November 2011) Gas chromatographic retention indices were evaluated for 505 frequently reported plant essential oil components using a large retention index database. Retention data are presented for three types of commonly used stationary phases: dimethyl silicone (nonpolar), dimethyl sili- cone with 5% phenyl groups (slightly polar), and polyethylene glycol (polar) stationary phases. The evaluations are based on the treatment of multiple measurements with the number of data records ranging from about 5 to 800 per compound. Data analysis was limited to temperature programmed conditions. The data reported include the average and median values of retention index with standard deviations and confidence intervals. VC 2011 by the U.S. Secretary of Commerce on behalf of the United States. All rights reserved. [doi:10.1063/1.3653552] Key words: essential oils; gas chromatography; Kova´ts indices; linear indices; retention indices; identification; flavor; olfaction. CONTENTS 1. Introduction The practical applications of plant essential oils are very 1. Introduction................................ 1 diverse. They are used for the production of food, drugs, per- fumes, aromatherapy, and many other applications.1–4 The 2. Retention Indices ........................... 2 need for identification of essential oil components ranges 3. Retention Data Presentation and Discussion . 2 from product quality control to basic research. The identifi- 4. Summary.................................. 45 cation of unknown compounds remains a complex problem, in spite of great progress made in analytical techniques over 5. -

The Composition of Strawberry Aroma Is Influenced by Cultivar, Maturity, and Storage Charles F
WORKSHOP The Composition of Strawberry Aroma Is Influenced by Cultivar, Maturity, and Storage Charles F. Forney1, Willy Kalt2, and Michael A. Jordan3 Agriculture and Agri-Food Canada, Atlantic Food and Horticulture Research Centre, 32 Main Street, Kentville, N.S., B4N 1J5, Canada Strawberry (Fragaria ×ananassa Duch.) fruit have a unique, they both may contribute to strawberry aroma (Dirinck et al., 1981; highly desirable flavor and are one of the most popular summer fruits. Schreier, 1980). Sugars, acids, and aroma volatiles contribute to the characteristic The volatile profile obtained from strawberry fruit is influenced by strawberry flavor, which is dependent on the proper balance of these the analytical methods used to characterize these compounds. Volatiles chemical constituents. While sugars and acids are responsible for the from whole, intact fruit can be collected using headspace techniques; sweetness and tartness of the fruit, aroma volatiles provide the unique, these samples can be analyzed directly or concentrated using adsor- fruity flavors that characterize a fresh strawberry. bent or cold traps. Volatiles are also collected from homogenized fruit The aroma of fresh strawberries is dependent on many factors. The or juice, using either headspace or solvent extraction techniques. large genetic variability in the nature of strawberry aroma results in Volatile samples are normally analyzed by gas liquid chromatography differences in flavor among cultivars. In addition, the aroma changes using a variety of methods of sample introduction, including liquid dramatically during fruit ripening after harvest; therefore, it is impor- injection, thermal desorption, and cold on-column injection. High tant to preserve and enhance the ripe fruit aroma during postharvest performance liquid chromatography (HPLC) has been used for some handling. -

Fermentation and Ester Taints
Fermentation and Ester Taints Anita Oberholster Introduction: Aroma Compounds • Grape‐derived –provide varietal distinction • Yeast and fermentation‐derived – Esters – Higher alcohols – Carbonyls – Volatile acids – Volatile phenols – Sulfur compounds What is and Esters? • Volatile molecule • Characteristic fruity and floral aromas • Esters are formed when an alcohol and acid react with each other • Few esters formed in grapes • Esters in wine ‐ two origins: – Enzymatic esterification during fermentation – Chemical esterification during long‐term storage Ester Formation • Esters can by formed enzymatically by both the plant and microbes • Microbes – Yeast (Non‐Saccharomyces and Saccharomyces yeast) – Lactic acid bacteria – Acetic acid bacteria • But mainly produced by yeast (through lipid and acetyl‐CoA metabolism) Ester Formation Alcohol function Keto acid‐Coenzyme A Ester Ester Classes • Two main groups – Ethyl esters – Acetate esters • Ethyl esters = EtOH + acid • Acetate esters = acetate (derivative of acetic acid) + EtOH or complex alcohol from amino acid metabolism Ester Classes • Acetate esters – Ethyl acetate (solvent‐like aroma) – Isoamyl acetate (banana aroma) – Isobutyl acetate (fruit aroma) – Phenyl ethyl acetate (roses, honey) • Ethyl esters – Ethyl hexanoate (aniseed, apple‐like) – Ethyl octanoate (sour apple aroma) Acetate Ester Formation • 2 Main factors influence acetate ester formation – Concentration of two substrates acetyl‐CoA and fusel alcohol – Activity of enzyme responsible for formation and break down reactions • Enzyme activity influenced by fermentation variables – Yeast – Composition of fermentation medium – Fermentation conditions Acetate/Ethyl Ester Formation – Fermentation composition and conditions • Total sugar content and optimal N2 amount pos. influence • Amount of unsaturated fatty acids and O2 neg. influence • Ethyl ester formation – 1 Main factor • Conc. of precursors – Enzyme activity smaller role • Higher fermentation temp formation • C and N increase small effect Saerens et al. -

Flavour in Food
Flavour in food Edited by Andree Voilley and Patrick Etievant d^^ CRC Press Boca Raton Boston New York Washington, DC WOODHEAD PUBLISHING LIMITEI Cambridge England Published by Woodhead Publishing Limited Abington Hall, Abington Cambridge CBl 6AH England www.woodheadpublishing.com Published in North America by CRC Press LLC 6000 Broken Sound Parkway, NW Suite 300 Boca Raton, FL 33487 USA First published 2006, Woodhead Publishing Limited and CRC Press LLC © 2006, Woodhead Publishing Limited The authors have asserted their moral rights. This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. Reasonable efforts have been made to publish reliable data and information, but the authors and the publishers cannot assume responsibility for the validity of all materials. Neither the authors nor the publishers, nor anyone else associated with this publication, shall be liable for any loss, damage or liability directly or indirectly caused or alleged to be caused by this book. Neither this book nor any part may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming and recording, or by any information storage or retrieval system, without permission in writing from Woodhead Publishing Limited. The consent of Woodhead Publishing Limited does not extend to copying for general distribution, for promotion, for creating new works, or for resale. Specific permission must be obtained in writing from Woodhead Publishing Limited for such copying. Trademark notice: Product or corporate names may be trademarks or registered trademarks, and are used only for identification and explanation, without intent to infringe. -

Final Report Research
Southern Region Small Fruit Consortium Final Report Research Title: Identifying Flavor and Aroma Attributes of Arkansas Fresh-Market Blackberries that Impact Marketability Grant Code: SRSFC Project # 2020-05 Grant Period: March 1, 2020-February 28, 2021 Name, Mailing and Email Address of Principal Investigator(s): Principal Investigator: Dr. Renee Threlfall, Research Scientist, Department of Food Science, 2650 N. Young Ave., University of Arkansas System Division of Agriculture, Fayetteville, AR 72704, [email protected] Co-Principal Investigators: Dr. John R. Clark, University Professor, Department of Horticulture, 316 Plant Science Building, University of Arkansas System Division of Agriculture, Fayetteville, AR 72701, [email protected] Dr. Margaret Worthington, Assistant Professor, Department of Horticulture, 316 Plant Science Building, University of Arkansas System Division of Agriculture, Fayetteville, AR 72701, [email protected] Dr. Luke Howard, Professor, Department of Food Science, 2650 N. Young Ave., University of Arkansas System Division of Agriculture, Fayetteville, AR 72704, [email protected] Public Abstract Fresh-market blackberries (Rubus subgenus Rubus) have unique aromas and flavors that appeal to consumers. Although the basic tastes (sweetness, sourness, and bitterness) impact the flavor perception of blackberries, volatile aroma compounds (substances which vaporize easily at ambient temperature) are also responsible for typical aromas and flavors. The berry attributes, composition attributes, and volatile aroma attributes of Arkansas fresh-market blackberries were measured. Blackberries were harvested from the University of Arkansas System Division of Agriculture Fruit Research Station, Clarksville, AR in June 2020. Nineteen genotypes were evaluated including nine cultivars (Caddo, Natchez, Osage, Ouachita, Ponca, Prime-Ark® 45, Prime-Ark® Horizon, Prime-Ark® Traveler, and Tupy) and ten breeding selections (A-2526T, A-2528T, A-2547T, A-2587T, A-2610T, A-2620T, A-2625T, A-2658T, A-2701T, and APF- 409T). -

B REGULATION (EC) No 1334/2008 of the EUROPEAN
2008R1334 — EN — 13.05.2016 — 011.001 — 1 This document is meant purely as a documentation tool and the institutions do not assume any liability for its contents ►B REGULATION (EC) No 1334/2008 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 December 2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods and amending Council Regulation (EEC) No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008 and Directive 2000/13/EC (Text with EEA relevance) (OJ L 354, 31.12.2008, p. 34) Amended by: Official Journal No page date ►M1 Commission Implementing Regulation (EU) No 872/2012 of 1 October L 267 1 2.10.2012 2012 ►M2 Commission Regulation (EU) No 545/2013 of 14 June 2013 L 163 15 15.6.2013 ►M3 Commission Regulation (EU) No 985/2013 of 14 October 2013 L 273 18 15.10.2013 ►M4 Commission Regulation (EU) No 246/2014 of 13 March 2014 L 74 58 14.3.2014 ►M5 Commission Regulation (EU) No 1098/2014 of 17 October 2014 L 300 41 18.10.2014 ►M6 Commission Regulation (EU) 2015/648 of 24 April 2015 L 107 15 25.4.2015 ►M7 Commission Regulation (EU) 2015/1102 of 8 July 2015 L 181 54 9.7.2015 ►M8 Commission Regulation (EU) 2015/1760 of 1 October 2015 L 257 27 2.10.2015 ►M9 Commission Regulation (EU) 2016/54 of 19 January 2016 L 13 40 20.1.2016 ►M10 Commission Regulation (EU) 2016/55 of 19 January 2016 L 13 43 20.1.2016 ►M11 Commission Regulation (EU) 2016/178 of 10 February 2016 L 35 6 11.2.2016 ►M12 Commission Regulation (EU) 2016/637 of 22 April 2016 L 108 24 23.4.2016 2008R1334 — EN — 13.05.2016 — 011.001 -

Monitoring Cider Aroma Development Throughout the Fermentation
beverages Article Monitoring Cider Aroma Development throughout the Fermentation Process by Headspace Solid Phase Microextraction (HS-SPME) Gas Chromatography–Mass Spectrometry (GC-MS) Analysis Matthew T. Bingman 1, Claire E. Stellick 1, Jordanne P. Pelkey 1, Jared M. Scott 2 and Callie A. Cole 1,* 1 Department of Chemistry and Biochemistry, Fort Lewis College, 1000 Rim Drive, Durango, CO 81301, USA; [email protected] (M.T.B.); [email protected] (C.E.S.); [email protected] (J.P.P.) 2 EsoTerra Cider, P.O. Box 156, Hesperus, CO 81326, USA; [email protected] * Correspondence: [email protected] Received: 19 April 2020; Accepted: 9 June 2020; Published: 16 June 2020 Abstract: Volatile organic compounds (VOCs) play a crucial role in cider quality. Many variables involved in the fermentation process contribute to cider fragrance, but their relative impact on the finished odor remains ambiguous, because there is little consensus on the most efficient method for cider volatile analysis. Herein, we have optimized and applied a headspace solid phase microextraction gas chromatography–mass spectrometry (HS-SPME GC-MS) method for the chemical analysis of cider VOCs. We determined that the 30 min exposure of a divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS) solid phase microextraction (SPME) fiber at 40 ◦C yielded detection of the widest variety of VOCs at an extraction efficiency >49% higher than comparable fibers. As a proof-of-concept experiment, we utilized this method to profile cider aroma development throughout the fermentation process for the first time. The results yielded a very practical outcome for cider makers: a pre-screening method for determining cider quality through the detection of off-flavors early in the fermentation process. -

Supplemental Material
Haddad Supp pp 1 Supplementary materials for: Global features of neural activity in the olfactory system form a parallel code that predicts olfactory behavior and perception Rafi Haddad1,2#, Tali Weiss1, Rehan Khan1σ, Boaz Nadler2, Nathalie Mandairon3, 3 1* 1*# Moustafa Bensafi , Elad Schneidman and Noam Sobel . Section 1: PCA analysis We provide a step by step example of how we conducted the PCA analysis. Assume we have 8 odors, each located on the vertexes of a 3 dimensional unit cube. Each odor can thus be represented by the exact binary code of the numbers 0 to 7. We can represent these odors in the following binary code matrix: Odor ID Pattern code: 1 0 0 0 2 0 0 1 3 0 1 0 4 0 1 1 5 1 0 0 6 1 0 1 7 1 1 0 8 1 1 1 1 Haddad Supp pp 2 Using any available mathematical tool (we used Matlab 'princomp' method) we can calculate the principle components scores of this matrix. In this case the values of the PC1 scores are: [1 0 0]. The PC1 projection value of each row is the projection of each row by the PC1 weight vector. For example the PC1 value of the first row is [0,0,0]X[1,0,0] = 0X1 + 0X0 + 0X0 = 0. (this is a vector multiplication). Thus the PC1 value of each row of this matrix is: 0,0,0,0,1,1,1,1. Note that usually the PC1 is calculated on a centered matrix (the columns of the matrix have zero mean) and thus the PC1 value might be shifted by some value. -
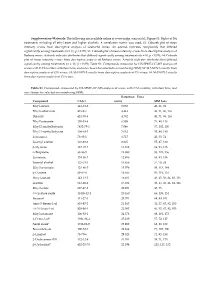
Supplementary File 1
Supplementary Materials: The following are available online at www.mdpi.com/xxx/s1, Figure S1: Biplot of BA treatments including all ethyl ester and higher alcohols. A correlation matrix was used; S2: Cobweb plot of mean intensity scores from descriptive analysis of Grenache wines. An asterisk indicates compounds that differed significantly among treatments (n = 11, p < 0.05); S3: Cobweb plot of mean intensity scores from descriptive analysis of Barbera wines. Asterisk indicates attributes that differed significantly among treatments (n = 10, p < 0.05); S4: Cobweb plot of mean intensity scores from descriptive analysis of Barbera wines. Asterisk indicates attributes that differed significantly among treatments (n = 10, p < 0.05); Table S1: Compounds measured by HS‐SPME‐GC‐MS analysis of wines with CAS number, retention time, and ions chosen for selected ion monitoring (SIM); S2: MANOVA results from descriptive analysis of GN wines; S3: MANOVA results from descriptive analysis of CS wines; S4: MANOVA results from descriptive analysis of CS wines. Table S1. Compounds measured by HS‐SPME‐GC‐MS analysis of wines with CAS number, retention time, and ions chosen for selected ion monitoring (SIM). Retention Time Compound CAS # (min) SIM Ions Ethyl acetate 141‐78‐6 3.053 43, 61, 88 Ethyl isobutyrate 97‐62‐1 4.443 43, 71, 86, 116 Diacetyl 431‐03‐8 4.782 43, 71, 86, 116 Ethyl butanoate 105‐54‐4 6.508 71, 88, 116 Ethyl 2‐methylbutyrate 7452‐79‐1 7.046 57, 102, 130 Ethyl 3‐methylbutyrate 108‐64‐5 7.632 55, 88, 130 Isobutanol 78‐83‐1 8.717 43, 55, -

Fermentation-Derived Aroma Compounds in Varietal Young Wines from South Africa
Fermentation-derived Aroma Compounds in Varietal Young Wines from South Africa L. Louw1 ' 2 , A.G.J. Tredoux1, P. Van Rensburg 1 ' 2 , M. Kidd3, T. Naes4 and H.H. Nieuwoudt 1 (1) Institute for Wine Biotechnology, Department of'Viticulture and Oenology, Stellenbosch University, Private Bag Xl, 7602, Matieland, South Africa (2)Distell Ltd., P.O. Box 184, Stellenbosch 7602, South Africa (3) Centre for Statistical Consultation, Stellenbosch University, Private Bag Xl, Matieland 7602, South Africa (4)Nofima Mat, Oslovegen 1, 1430 As, Norway Submitted for publication: May 2010 Accepted for publication: September 2010 Key words: South African young wines, GC-FID, aroma compounds, odour activity, discriminant analysis The volatile composition of 925 single cultivar young Sauvignon blanc, Chardonnay, Pinotage, Merlot, Shiraz and Cabernet Sauvignon wines of vintages 2005 to 2007, was determined using gas chromatography - flame ionisation detection. Compositional data were compared to published data on young wines from South Africa and other countries. South African young wines analysed in this study had a largely similar volatile composition to that reported in the literature. Significant between-vintage and between-cultivar differences were observed in the volatile composition of the wines investigated in this study. The concentration ranges of four compounds in red wines, hexanol, propanol, diethyl succinate and ethyl lactate, and four compounds in white wines, 2-phenylethanol, hexanoic acid, isoamyl acetate and propanol, were not influenced by vintage effects. This finding was interpreted as the first indication that typical concentration ranges for some aroma compounds can be established for South African young cultivar wines. A trend was observed in the white wines that the alcohols and their respective acetate esters, as well as fatty acids and their ethyl esters, were responsible for the vintage-related effects. -

Analysis of Odourant Compounds in Wine - with Headspace Solid-Phase Microextraction and Gas Chromatography- Mass Spectrometry
Analysis of odourant compounds in wine - With headspace solid-phase microextraction and gas chromatography- mass spectrometry By Emma Ödmar Analytical science program in chemistry with focus on forensics Candidate for Degree of Bachelor of Science School of Science and Technology Örebro university Spring term 2018 Table of contents Abstract .................................................................................................................................. 3 1. Introduction......................................................................................................................... 4 1.1 Aim ............................................................................................................................... 4 1.2 Scope ............................................................................................................................. 4 1.3 Background ................................................................................................................... 4 1.3.1 Wine ....................................................................................................................... 4 1.3.2 Headspace solid-phase microextraction (HS-SPME)................................................ 5 1.3.3 Gas chromatography-mass spectrometry (GC-MS) .................................................. 6 2. Materials and methods......................................................................................................... 7 2.1 Materials....................................................................................................................... -

Download This Article in PDF Format
BIO Web of Conferences 15, 02032 (2019) https://doi.org/10.1051/bioconf/20191502032 42nd World Congress of Vine and Wine Influence of enzymatic treatments on white wine composition E.C. Scutaras¸u1, V.V. Cotea1, C.E. Luchian1, L.C. Colibaba2, N. Katalin2, R. Oprean3, and M. Niculaua 1 University of Agricultural Sciences and Veterinary Medicine “Ion Ionescu de la Brad”, Ias¸i, Romania 2 “Iuliu Has¸ieganu” University of Medicine and Pharmacy, V. Babes¸ Street, Cluj Napoca, Romania 3 Research Center for Oenology, Romanian Academy – Ias¸i Branch, Romania Abstract. Most biochemical reactions involved in the wine-making process are catalyzed by enzymes. The use of enzymes of exogenous origin in wine production is due to the numerous technological and economical advantages demonstrated over time in the winemaking process. Understanding the important role played by enzymes in wine making technology contributes to the development of optimization strategies for the production process to improve the final quality of the wine. In order to accomplish this study, the influence of five oenological preparations with pectolytic and β-glucosidases enzymes types on the volatile compounds of white wines obtained from Feteascaregal˘ a˘ variety was analyzed by monitoring their evolution during the alcoholic fermentation to the final product. Wine samples have been physically and chemically analysed (pH, acidity, alcoholic strength, density, malic acid, lactic acid, sugar content, SO2,total dry extract and non-reducing extract) according to OIV Standards. Separation and identification of flavor compounds was performed using an Agilent 7890 gas chromatograph coupled to a 5975 C inert XL EI/CI MSD spectrophotometer.