In Crop Science and Horticulture
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Plantlist Master 09 15 09
Bareroot Fruit,Trees, Shrubs, and Vines 2010 Fruit Harcot Apricot Excellent flavor and quality. Large, sweet, juicy, rich Apples flavor. Brown rot resistant. Mid-June. Trees range from dwarf to very large. Most are narrow and upright. White to pale pink flowers are showy in late March - early April. Fruit on short spurs which take 3 - 4 years to form. Cherries Prune for size control, remove suckers and crossing Very upright growing trees to 10’ - 30’. Can be trained branches. smaller, but avoid heavy pruning of mature trees. Tolerant of heavy soils, drought, or lawn watering. Showy pure white flowers in March. Braeburn Apple Fruit on short spurs, which take 3 - 4 years to form. Prune only for size control, if at all. Late season, crisp and tangy, similar to Granny Smith Moderately tolerant of drought. No lawn watering. but richer flavor. Excellent keeper. Green with dark red blush. October-November harvest. Self-fruitful. Bada Bing Cherry Self-fruitful version of the commercial sweet cherry. Late May - early June harvest. Fuji Apple Sweet, crisp and flavorful, excellent keeper. Late September. Excellent pollenizer for other apple varieties. Self-fruitful. Lapins Cherry Large dark red, firm, sweet fruit like Bing. “Self- fertile Bing”. Resists cracking. Early to mid June. Granny Smith Apple Bright green skin, tart/sweet flavor, great texture. For eating, cooking, sauce. October - November harvest. Rainier Cherry Yellow with red blush. Very sweet, fine texture, very firm. Resists cracking. Pollenizer needed. Late May to Red Gravenstein Apple early June. Red variant of the applesauce apple, also great for cooking. Fine for fresh eating if picked a little underripe. -

Title Molecular and Biochemical Studies on Self-Incompatibility of Two Commercially Important Cherry Species (Prunus Avium L. An
Molecular and biochemical studies on self-incompatibility of Title two commercially important cherry species (Prunus avium L. and P. cerasus L.)( Dissertation_全文 ) Author(s) Yamane, Hisayo Citation 京都大学 Issue Date 2003-01-23 URL https://doi.org/10.14989/doctor.r11106 Right Type Thesis or Dissertation Textversion author Kyoto University 859 Molecular and Biochemical Studies on Self-incompatibility of Two Commercially Important Cherry Species (Prunus avium L. and P. cerasus L.) HISAYO YAMANE Molecular and Biochemical Studies on Self-incompatibility of Two Commercially Important Cherry Species (Prunus avium L. and P. cerasus L.) HISAYO YAMANE Contents General Introduction ------------------------------------------------------------------------------- 1 Chapter 1. Identification and characterization of S-RNases associated with gametophytic self-incompatibility in sweet cherry (Prunus avium L.) 1.1. Introduction -------------------------------------------------------------------- 5 1.2. Materials and Methods ------------------------------------------------------- 5 1.3. Results and Discussion ------------------------------------------------------- 11 1.4. Summary ----------------------------------------------------------------------- 26 Chapter 2. Molecular typing of S-haplotypes in sweet cherry (Prunus avium L.) 2.1. Introduction -------------------------------------------------------------------- 27 2.2. Revisiting the S-haplotype nomenclature of sweet cherry by RFLP analysis 2.2.1. Materials and Methods -------------------------------------------- -

SWEET CHERRIES: Production, Marketing, and Processing
árf-3 SWEET CHERRIES: Production, Marketing, and Processing Agriculture Handbook No. 442 p^ C_^ '"^ CI? -j-, ■ —Í 00 ::^ Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE PRECAUTION Pesticides used improperly can be injurious to man, animals, and plants. Follow the directions and need all precautions on the labels. Store pesticides in original containers under lock and key—out of the reach of children and animals—and away from food and feed. Apply pesticides so that they do not endanger humans, livestock, crops, beneficial insects, fish, and wildlife. Do not apply pesticides when there is danger of drift, when honey bees or other' pol- linating insects are visiting plants, or in ways that may contaminate water or leave illegal resi- dues. Avoid prolonged inhalation of pesticide sprays or dusts; wear protective clothing and equipment if specified on the container. If your hands become contaminated with a pesticide, do not eat or drink until you have washed In case a pesticide is swallowed or gets in the eyes, follow the first aid treatment given on the label, and get prompt medical attention. If a pesticide is spilled on your skin or clothing, remove clothing immediately and wash skin thoroughly. Do not clean spray equipment or dump excess spray material near ponds, streams, or wells Be- cause It is difficult to remove all traces of herbicides from equipment, do not use the same equip- ment for insecticides or fungicides that you use for herbicides. Dispose of empty pesticide containers promptly. Have them buried at a sanitary land-fill dump or crush and bury them in a level, isolated place. -

Bareroot 2021
El Dorado Nursery and Garden BAREROOT LIST 2021 (8-12-20) CHILLING HOURS DEFINED: Chilling refers to the number of hours, 45 degrees F and under, during the dormancy period. All fruit and nut trees need a specific amount of chilling hours before they will produce fruit. The amount varies with each variety and the hours need not be continuous. In the description of the fruit tree, 800 hours (for example) means it needs that many minimum hours of chilling time. Special Order Fruit Trees (SOFT) Must be ordered by November 1, 2020 There is a small additional charge for these varieties. Deposit required, must be picked up from Nursery by February 15, 2021. Deposit refunded only if there is a crop failure. ** No discounts ** APPLE COX ORANGE PIPPIN APPLE Old favorite dessert apple. Firm, juicy, sweet, rich flavor, not tart. Distinctive aroma. Skin is orange-red to bright red over yellow. Prefers moderate climate. AKANE APPLE Especially nice red dessert apple derived from Mid-season. 800 hours. Self-fruitful. Jonathan - sweet, rich, spicy flavor. Resists scab and powdery mildew. Harvest in early season (Aug in Central Cal). 800 hours. DOLGO CRABAPPLE Long-time favorite all-purpose crabapple. Pollinizer required. Large, fragrant white flowers. Fruit makes tasty, bright-red jelly. Vigorous, upright, open tree to 30 feet x 25 feet, depending on ASHMEAD’S KERNEL Many think of this apple as one of the conditions. Resistant to scab, rust, mildew, fireblight. 500 hours. all-time best flavored. Small to medium fruit, used for dessert, Self-fruitful. cider, and sauce. Good keeper. -

Fruit Trees & Small Fruits
FRUIT TREES & SMALL FRUITS Apple 'Cortland' Semi-Dwarf Cortland Apple 12' x 12' white Apple 'Cortland' semi-dwarf #5 FP $56.98 Apple 'Empire' Semi-Dwarf Empire Apple 12' x 12' white Apple 'Empire' #7 5-7' $79.98 Apple 'Fuji' Semi-Dwarf Fuji Apple 12' x 12' white Apple 'Fuji' semi-dwarf #5 $79.98 Apple 'Granny Smith' Semi-Dwarf Granny Smith Apple 12' x 12' white Apple 'Granny Smith' #5 $79.98 Apple 'Honeycrisp' Honeycrisp Semi Dwarf Apple 12' x 12' white Apple 'Honeycrisp' semi-dwarf #5 FP $56.98 Apple 'Honeycrisp' semi-dwarf #7 $79.98 Apple 'McIntosh' Semi-Dwarf MacIntosh Apple 12' x 12' white Apple 'McIntosh' semi-dwarf #5 FP $56.98 Apple 'Northern Spy' Semi-Dwarf Northern Spy Apple 12' x 12' white Apple 'Northern Spy' #7 5-7' $79.98 Apple 'Red Delicious' Semi-Dwarf Red Delicious Apple 12' x 12' white Apple 'Red Delicious' semi-dwarf #5 FP $56.98 Apple 'Royal Gala' Semi-Dwarf Royal Gala Apple 12' x 12' white Apple 'Royal Gala' #7 5-7' $79.98 Apple 'Yellow Delicious' Semi-Dwarf Yellow Dellicious Apple 12' x 12' white Apple 'Yellow Delicious' semi-dwarf #5 FP $56.98 Apple 'Yellow Delicious' semi-dwarf #5 $79.98 Apricot 'Goldcot' Goldcot Apricot 15' x 15' white Apricot 'Goldcot' #7 $79.98 Apricot 'Tomcot' Semi-Dwarf Tomcot Apricot 17' x 17' white Blackberry 'Arapaho' #1 $17.98 Blackberry 'Arapaho' #3 $44.98 Blueberry 'Blue Jay' Blue Jay Blueberry 6' x 6' white Blueberry 'Blue Jay' AB #3 $52.98 Blueberry 'Blueberry Glaze' Blueberry Glaze Blueberry 2.5' x 2.5' white Blueberry 'Blueberry Glaze' #19cm $29.98 Blueberry 'Blueberry Glaze' #2 $46.98 -

Genetic Diversity and Relatedness of Sweet Cherry (Prunus Avium L.) Cultivars Based on Single Nucleotide Polymorphic Markers
ORIGINAL RESEARCH ARTICLE published: 25 June 2012 doi: 10.3389/fpls.2012.00116 Genetic diversity and relatedness of sweet cherry (Prunus avium L.) cultivars based on single nucleotide polymorphic markers Angel Fernandez i Marti 1,2, Blessing Athanson3,Tyson Koepke 4, Carolina Font i Forcada5, Amit Dhingra4 and Nnadozie Oraguzie 3* 1 Departamento Biología Molecular, Parque Científico Tecnológico Aula Dei, Zaragoza, Spain 2 Unidad de Fruticultura, Centro de Investigación y Tecnología Agroalimentario de Aragón, Zaragoza, Spain 3 Irrigated Agriculture Research and Extension Centre, Washington State University, Pullman, WA, USA 4 Department of Horticulture and Landscape Architecture, Washington State University, Pullman, WA, USA 5 Departamento de Pomología, Estación Experimental de Aula Dei (CSIC), Zaragoza, Spain Edited by: Most previous studies on genetic fingerprinting and cultivar relatedness in sweet cherry Sun Hee Woo, Chungbuk National were based on isoenzyme, RAPD, and simple sequence repeat (SSR) markers. This study University, South Korea was carried out to assess the utility of single nucleotide polymorphism (SNP) markers Reviewed by: Md. Shafikur Rahman, Patuakhali generated from 3 untranslated regions (UTR) for genetic fingerprinting in sweet cherry. A Science and Technology University, total of 114 sweet cherry germplasm representing advanced selections, commercial cul- Bangladesh tivars, and old cultivars imported from different parts of the world were screened with Abu Hena Mostafa Kamal, Korea seven SSR markers developed from other Prunus species and with 40 SNPs obtained Research Institute of Bioscience and Biotechnology, South Korea from 3 UTR sequences of Rainier and Bing sweet cherry cultivars. Both types of marker *Correspondence: study had 99 accessions in common. The SSR data was used to validate the SNP results. -
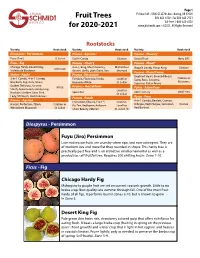
Fruit Trees for 2020-2021
Page 1 P O Box 189 • 9500 SE 327th Ave • Boring, OR 97009 Fruit Trees 503-663-4128 • Fax 503-663-2121 Toll-Free 1-800-825-8202 for 2020-2021 www.jfschmidt.com • ©2021, All Rights Reserved Rootstocks Variety Rootstock Variety Rootstock Variety Rootstock Diospyros - Persimmon Prunus - Aprium® Prunus - Pluerry® Fuyu (‘Jiro’) D. Lotus Cot-N-Candy Citation Sweet Treat Myro 29C Ficus - Fig Prunus - Cherry Prunus - Pluot® Chicago Hardy, Desert King, 4-in-1, Bing, Montmorency, Mahaleb or own root Dapple Dandy, Flavor King Citation Violette de Bordeaux Rainier, Stella, Utah Giant, Van Mazzard Prunus - Plum/Prune Malus - Apple Prunus - Nectarine Elephant Heart, Emerald Beaut, Citation or 3-in-1 Combo, 4-in-1 Combo, Fantasia, Flavortop, Harko, Lovell or Santa Rosa, Satsuma, Marianna Braeburn, Fuji, Gala, Ghost, Heavenly White St. Julian Superior, Italian Prune Golden Delicious, Granny M106 Prunus - NectaPlum® Pyrus - Asian Pear Smith, Gravenstein, Honeycrisp, Lovell or Spice Zee 20th Century OHxF 333 Hudson's Golden Gem, Pink St. Julian Lady, McIntosh, Red Delicious Prunus - Peach Pyrus - Pear Prunus - Apricot Contender, Elberta, Frost™, Citation, 4-in-1 Combo, Bartlett, Comice, Harcot, Perfection, Tilton, Citation or Pix Zee, Redhaven, Reliance Lovell or D’Anjou, Red D’Anjou, Sensation Quince Wenatchee Moorpark St. Julian Snow Beauty, Veteran St. Julian “A” Red Bartlett Diospyros - Persimmon Fuyu (Jiro) Persimmon Late midseason fruits are crunchy when ripe, and non-astringent. They are of medium size and more flat than rounded in shape. This hardy tree is practically pest free and is an attractive small ornamental as well as a productive, self-fruitful tree. -

Adams Gardens & Nursery 4500 Greenhurst Road Nampa, Idaho
Adams Gardens & Nursery 4500 Greenhurst Road Nampa, Idaho 83686 208.461.6101 Plant varieties ordered from Dave Wilson Nursery for 2017: Gala Apple Wonderful dessert apple from New Zealand. Crisp, nice blend of sweetness and tartness, rich flavor. Skin reddish orange over yellow. Early harvest, 2 - 3 weeks before Red Delicious. Good pollenizer for other varieties. Adapted to cold- and warm-winter climates. Chilling requirement less than 500 hours. Self-fruitful. USDA Zones 4-10. Golden Delicious Apple Long-time favorite for its sweetness and flavor. Reliable producer, adapted to many climates. Pollenizer for Red Delicious. Midseason harvest (September in Central CA). 700 hours. Self-fruitful. USDA Zones 5-10. Granny Smith Apple From New Zealand. Large, late, green, all-purpose. Crisp, tart, eXcellent keeper. Requires long summer. Thrives in hot climates. 400 hours. Prolonged bloom: good pollenizer for other apples. Self-fruitful. USDA Zones 6-9. Honeycrisp Apple Winter hardy tree from the University of Minnesota. Fruit is crisp and juicy with an aromatic flavor. Striped red over yellow color. Stores well. Ripens mid-August. Pollenized by Gala, Granny Smith, Empire, McIntosh and Red Delicious. USDA Zones 3-8. McIntosh Apple Round, bright to dark red over green, superb quality in cool climates. Crisp, aromatic, subacid & sweet. Dessert/cooking. Early harvest. 900 hours. Partly self-fruitful, or pollenized by Red Delicious, Gala, or other. USDA Zones 4-7. Multi-Bud Apple, Fuji-Gala-Golden Delicious-Granny Smith Fuji, Gala, Golden Delicious and Granny Smith budded onto M-111 rootstock. Finished trees include 4n1's plus assorted 3n1's and 2n1's. -

PEF) Processing on the Physical, Chemical and Microbiological Properties of Red-Fleshed Sweet Cherries (Prunus Avium Var
Effects of Pulsed Electric Field (PEF) processing on the physical, chemical and microbiological properties of Red-Fleshed Sweet Cherries (Prunus avium var. Stella) By Kristine Ann Gualberto Sotelo A thesis submitted to Auckland University of Technology in partial fulfilment of the requirements for the degree of Master of Science (MSc) May 2015 School of Applied Sciences Primary Supervisor: Dr. Nazimah Hamid Secondary Supervisors: Dr. Noemi Gutierrez-Maddox Dr. Chris Pook Abstract The effects of mild or moderate intensity pulsed electric field (PEF) processing on the chemical, flavor and microbiological properties of Stella sweet cherries were studied. The specific aims of this study were to determine (1) the effect of different PEF treatments on the physico-chemical properties of cherry samples, (2) the effects of different PEF treatments and storage conditions on the volatile profile of cherry samples, (3) the effects of different PEF treatments and storage conditions on the extraction or release of bioactive compounds including the anthocyanins and polyphenols on PEF-treated samples, and finally (4) the growth of probiotic bacteria specifically the lactic acid bacteria on PEF-treated cherry samples, The cherry samples were treated at a constant pulse frequency of 100 Hz and a constant pulse width of 20 μs with different electric field strengths between 0.3 and 2.5 kV/cm used. Cherry fruits used in this study that were subjected to PEF processing were analysed (1) immediately after PEF (S2) and (2) 24 hours of incubation at 4 °C (S3). PEF treated samples were significantly different to control sample in terms of juice yield, pH, titratable acidity (TA), total soluble solids (TSS) and moisture content (MC). -

Fabulous Fruits for Front Range Gardeners
FABULOUS FRUITS FOR FRONT RANGE GARDENERS Craig R. Miller Parks & Open Space Manager www.cpnmd.org Selection of Fruit Trees . Select Colorado hardy varieties of fruit trees. Some varieties will not tolerate low winter temperatures and are more susceptible to fire blight. Peach, apricot and sweet cherry trees are sensitive to spring frosts and will not always produce a bountiful crop. Apricots rarely fruit here because of late frosts in April, but are worthwhile landscape plants thanks to their fall color, summer foliage and other ornamental qualities. Peaches also are not dependable, but more so than apricots, as they flower later. Pollination of Fruit Trees . Apples – In general, apple trees need another variety to be productive. That variety needs to be blooming at the same time and be within a distance of ¼ mile (the distance a bee can fly). In the Denver Metro area, there are enough of both apple and crabapple trees that pollination is usually not a problem. But if you want to play it safe, plant 2 varieties within 100 feet of each other. Apricots – Use two varieties. Nanking cherry will also do the job. Sweet Cherries – Use two varieties. Sour cherries will also work as a pollinator. Sour Cherries – Self-fertile. Peaches – Self fertile. Pears – Most need another variety for a better yield. Ornamental pears will work as well. Plums – Most need two varieties. American plum or western sand cherry will also work. Pruning & Thinning Fruit Trees . Dwarf trees are easily trained with little pruning. If pruning is needed, save fruit-bearing branches and eliminate diseased, dead, thin or rubbing branches. -

Fruit Trees Like
One of life’s greatest pleasures is biting into a fresh fruit – especially when it’s one you grew! Fruit trees like: A Sunny Location ● Good Soil ● Regular Watering Nectarine Hosui Asian Pear ‘Fantasia’ Round, brownish- Red/yellow fruit in orange fruit in fall mid-summer; large freestone, firm yellow flesh Bartlett Pear Red Bartlett Pear Medium to large Juicy red, sweet & juicy yellow, fragrant tender fruit in late fruit in late summer; summer eat fresh or can Bosc Pear Granny Smith Russet brown, juicy Apple fruit in fall with white Green, tasty apples flesh; good for in early fall eating, drying or canning Gravenstein Apple Fuji Apple Striped green, juicy Excellent fruit that apples in late keeps well. Top summer; great fresh quality apples in or cooked late fall Gala Apple Red Delicious Blushed yellow Apple striped apples in late Juicy, red sweet summer; semi— apples in late fall; sweet apples that very flavorful store well Blenheim Apricot Tilton Apricot Orange/yellow Yellow, tasty sweet apricots in apricots in mid early summer – use summer; smallish fresh or can and can well Elberta Peach Pluot Flavor King Blushed yellow Sweet flavor fruit in peaches in mid- late August summer Rio Oso Gem Satsuma Plum Peach Sweet, red juicy Sweet, rich-flavored, plums in mid- large, yellow summer peaches in August Babcock Peach Elephant Heart Pink, juicy peaches Plum in mid-summer Flavorful red, juicy plums in mid summer Frost Peach Burgundy Plum Yellow with red Sweet, deep red blush in late spring – flesh in early July resistant to peach- leaf curl Red Haven Peach Santa Rosa Plum Red/yellow juicy Deep red tasty peaches in mid plums in early summer summer Bing Cherry Lapins Cherry Dark red, juicy Black cherry in late cherries in mid summer – sweet summer – great cherry excellent in canned or frozen flavor and texture Black Tartarian Stella Cherry Cherry Firm and large Purplish/black sweet black, sweet cherries in early cherries in mid summer summer Rainier Sweet Cherry Large, sweet yellow fruit in mid summer . -

Genetic Diversity and Relatedness of Sweet Cherry (Prunus Avium L.) Cultivars Based on Single Nucleotide Polymorphic Markers
ORIGINAL RESEARCH ARTICLE published: 25 June 2012 doi: 10.3389/fpls.2012.00116 Genetic diversity and relatedness of sweet cherry (Prunus avium L.) cultivars based on single nucleotide polymorphic markers Angel Fernandez i Marti 1,2, Blessing Athanson3,Tyson Koepke 4, Carolina Font i Forcada5, Amit Dhingra4 and Nnadozie Oraguzie 3* 1 Departamento Biología Molecular, Parque Científico Tecnológico Aula Dei, Zaragoza, Spain 2 Unidad de Fruticultura, Centro de Investigación y Tecnología Agroalimentario de Aragón, Zaragoza, Spain 3 Irrigated Agriculture Research and Extension Centre, Washington State University, Pullman, WA, USA 4 Department of Horticulture and Landscape Architecture, Washington State University, Pullman, WA, USA 5 Departamento de Pomología, Estación Experimental de Aula Dei (CSIC), Zaragoza, Spain Edited by: Most previous studies on genetic fingerprinting and cultivar relatedness in sweet cherry Sun Hee Woo, Chungbuk National were based on isoenzyme, RAPD, and simple sequence repeat (SSR) markers. This study University, South Korea was carried out to assess the utility of single nucleotide polymorphism (SNP) markers Reviewed by: Md. Shafikur Rahman, Patuakhali generated from 3 untranslated regions (UTR) for genetic fingerprinting in sweet cherry. A Science and Technology University, total of 114 sweet cherry germplasm representing advanced selections, commercial cul- Bangladesh tivars, and old cultivars imported from different parts of the world were screened with Abu Hena Mostafa Kamal, Korea seven SSR markers developed from other Prunus species and with 40 SNPs obtained Research Institute of Bioscience and Biotechnology, South Korea from 3 UTR sequences of Rainier and Bing sweet cherry cultivars. Both types of marker *Correspondence: study had 99 accessions in common. The SSR data was used to validate the SNP results.