Using Raxml-NG in Practice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 A) Login to the System B) Use the Appropriate Command to Determine Your Login Shell C) Use the /Etc/Passwd File to Verify the Result of Step B
CSE ([email protected] II-Sem) EXP-3 1 a) Login to the system b) Use the appropriate command to determine your login shell c) Use the /etc/passwd file to verify the result of step b. d) Use the ‘who’ command and redirect the result to a file called myfile1. Use the more command to see the contents of myfile1. e) Use the date and who commands in sequence (in one line) such that the output of date will display on the screen and the output of who will be redirected to a file called myfile2. Use the more command to check the contents of myfile2. 2 a) Write a “sed” command that deletes the first character in each line in a file. b) Write a “sed” command that deletes the character before the last character in each line in a file. c) Write a “sed” command that swaps the first and second words in each line in a file. a. Log into the system When we return on the system one screen will appear. In this we have to type 100.0.0.9 then we enter into editor. It asks our details such as Login : krishnasai password: Then we get log into the commands. bphanikrishna.wordpress.com FOSS-LAB Page 1 of 10 CSE ([email protected] II-Sem) EXP-3 b. use the appropriate command to determine your login shell Syntax: $ echo $SHELL Output: $ echo $SHELL /bin/bash Description:- What is "the shell"? Shell is a program that takes your commands from the keyboard and gives them to the operating system to perform. -
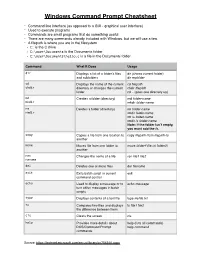
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

Don't Trust Traceroute (Completely)
Don’t Trust Traceroute (Completely) Pietro Marchetta, Valerio Persico, Ethan Katz-Bassett Antonio Pescapé University of Southern California, CA, USA University of Napoli Federico II, Italy [email protected] {pietro.marchetta,valerio.persico,pescape}@unina.it ABSTRACT In this work, we propose a methodology based on the alias resolu- tion process to demonstrate that the IP level view of the route pro- vided by traceroute may be a poor representation of the real router- level route followed by the traffic. More precisely, we show how the traceroute output can lead one to (i) inaccurately reconstruct the route by overestimating the load balancers along the paths toward the destination and (ii) erroneously infer routing changes. Categories and Subject Descriptors C.2.1 [Computer-communication networks]: Network Architec- ture and Design—Network topology (a) Traceroute reports two addresses at the 8-th hop. The common interpretation is that the 7-th hop is splitting the traffic along two Keywords different forwarding paths (case 1); another explanation is that the 8- th hop is an RFC compliant router using multiple interfaces to reply Internet topology; Traceroute; IP alias resolution; IP to Router to the source (case 2). mapping 1 1. INTRODUCTION 0.8 Operators and researchers rely on traceroute to measure routes and they assume that, if traceroute returns different IPs at a given 0.6 hop, it indicates different paths. However, this is not always the case. Although state-of-the-art implementations of traceroute al- 0.4 low to trace all the paths -

Forest Quickstart Guide for Linguists
Forest Quickstart Guide for Linguists Guido Vanden Wyngaerd [email protected] June 28, 2020 Contents 1 Introduction 1 2 Loading Forest 2 3 Basic Usage 2 4 Adjusting node spacing 4 5 Triangles 7 6 Unlabelled nodes 9 7 Horizontal alignment of terminals 10 8 Arrows 11 9 Highlighting 14 1 Introduction Forest is a package for drawing linguistic (and other) tree diagrams de- veloped by Sašo Živanović. This manual provides a quickstart guide for linguists with just the essential things that you need to get started. More 1 extensive documentation is available from the CTAN-archive. Forest is based on the TikZ package; more information about its commands, in par- ticular those controlling the appearance of the nodes, the arrows, and the highlighting can be found in the TikZ documentation. 2 Loading Forest In your preamble, put \usepackage[linguistics]{forest} The linguistics option makes for nice trees, in which the branches meet above the two nodes that they join; it will also align the example number (provided by linguex) with the top of the tree: (1) CP C IP I VP V NP 3 Basic Usage Forest uses a familiar labelled brackets syntax. The code below will out- put the tree in (1) above (\ex. requires the linguex package and provides the example number): \ex. \begin{forest} [CP[C][IP[I][VP[V][NP]]]] \end{forest} Forest will parse the above code without problem, but you are likely to soon get lost in your labelled brackets with more complicated trees if you write the code this way. The better alternative is to arrange the nodes over multiple lines: 2 \ex. -

NETSTAT Command
NETSTAT Command | NETSTAT Command | Use the NETSTAT command to display network status of the local host. | | ┌┐────────────── | 55──NETSTAT─────6─┤ Option ├─┴──┬────────────────────────────────── ┬ ─ ─ ─ ────────────────────────────────────────5% | │┌┐───────────────────── │ | └─(──SELect───6─┤ Select_String ├─┴ ─ ┘ | Option: | ┌┐─COnn────── (1, 2) ──────────────── | ├──┼─────────────────────────── ┼ ─ ──────────────────────────────────────────────────────────────────────────────┤ | ├─ALL───(2)──────────────────── ┤ | ├─ALLConn─────(1, 2) ────────────── ┤ | ├─ARp ipaddress───────────── ┤ | ├─CLients─────────────────── ┤ | ├─DEvlinks────────────────── ┤ | ├─Gate───(3)─────────────────── ┤ | ├─┬─Help─ ┬─ ───────────────── ┤ | │└┘─?──── │ | ├─HOme────────────────────── ┤ | │┌┐─2ð────── │ | ├─Interval─────(1, 2) ─┼───────── ┼─ ┤ | │└┘─seconds─ │ | ├─LEVel───────────────────── ┤ | ├─POOLsize────────────────── ┤ | ├─SOCKets─────────────────── ┤ | ├─TCp serverid───(1) ─────────── ┤ | ├─TELnet───(4)───────────────── ┤ | ├─Up──────────────────────── ┤ | └┘─┤ Command ├───(5)──────────── | Command: | ├──┬─CP cp_command───(6) ─ ┬ ────────────────────────────────────────────────────────────────────────────────────────┤ | ├─DELarp ipaddress─ ┤ | ├─DRop conn_num──── ┤ | └─RESETPool──────── ┘ | Select_String: | ├─ ─┬─ipaddress────(3) ┬ ─ ───────────────────────────────────────────────────────────────────────────────────────────┤ | ├─ldev_num─────(4) ┤ | └─userid────(2) ─── ┘ | Notes: | 1 Only ALLCON, CONN and TCP are valid with INTERVAL. | 2 The userid -

Introduction to Unix Shell
Introduction to Unix Shell François Serra, David Castillo, Marc A. Marti- Renom Genome Biology Group (CNAG) Structural Genomics Group (CRG) Run Store Programs Data Communicate Interact with each other with us The Unix Shell Introduction Interact with us Rewiring Telepathy Typewriter Speech WIMP The Unix Shell Introduction user logs in The Unix Shell Introduction user logs in user types command The Unix Shell Introduction user logs in user types command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off shell The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output -

Linux Terminal Commands Man Pwd Ls Cd Olmo S
Python Olmo S. Zavala Romero Welcome Computers File system Super basics of Linux terminal Commands man pwd ls cd Olmo S. Zavala Romero mkdir touch Center of Atmospheric Sciences, UNAM rm mv Ex1 August 9, 2017 Regular expres- sions grep Ex2 More Python Olmo S. 1 Welcome Zavala Romero 2 Computers Welcome 3 File system Computers File system 4 Commands Commands man man pwd pwd ls ls cd mkdir cd touch mkdir rm touch mv Ex1 rm Regular mv expres- sions Ex1 grep Regular expressions Ex2 More grep Ex2 More Welcome to this course! Python Olmo S. Zavala Romero Welcome Computers File system Commands 1 Who am I? man pwd 2 Syllabus ls cd 3 Intro survey https://goo.gl/forms/SD6BM6KHKRlDOpZx1 mkdir 4 touch Homework 1 due this Sunday. rm mv Ex1 Regular expres- sions grep Ex2 More How does a computer works? Python Olmo S. Zavala Romero Welcome Computers File system Commands man pwd ls cd mkdir touch rm mv Ex1 Regular 1 CPU Central Processing Unit. All the computations happen there. expres- sions 2 HD Hard Drive. Stores persistent information in binary format. grep 3 RAM Random Access Memory. Faster memory, closer to the CPU, not persistent. Ex2 More 4 GPU Graphics Processing Unit. Used to process graphics. https://teentechdaily.files.wordpress.com/2015/06/computer-parts-diagram.jpg What is a file? Python Olmo S. Zavala Romero Welcome Computers File system Commands man pwd ls cd mkdir touch rm mv Section inside the HD with 0’s and 1’s. Ex1 Regular File extension is used to identify the meaning of those 0’s and 1’s. -

Freebsd Command Reference
FreeBSD command reference Command structure Each line you type at the Unix shell consists of a command optionally followed by some arguments , e.g. ls -l /etc/passwd | | | cmd arg1 arg2 Almost all commands are just programs in the filesystem, e.g. "ls" is actually /bin/ls. A few are built- in to the shell. All commands and filenames are case-sensitive. Unless told otherwise, the command will run in the "foreground" - that is, you won't be returned to the shell prompt until it has finished. You can press Ctrl + C to terminate it. Colour code command [args...] Command which shows information command [args...] Command which modifies your current session or system settings, but changes will be lost when you exit your shell or reboot command [args...] Command which permanently affects the state of your system Getting out of trouble ^C (Ctrl-C) Terminate the current command ^U (Ctrl-U) Clear to start of line reset Reset terminal settings. If in xterm, try Ctrl+Middle mouse button stty sane and select "Do Full Reset" exit Exit from the shell logout ESC :q! ENTER Quit from vi without saving Finding documentation man cmd Show manual page for command "cmd". If a page with the same man 5 cmd name exists in multiple sections, you can give the section number, man -a cmd or -a to show pages from all sections. man -k str Search for string"str" in the manual index man hier Description of directory structure cd /usr/share/doc; ls Browse system documentation and examples. Note especially cd /usr/share/examples; ls /usr/share/doc/en/books/handbook/index.html cd /usr/local/share/doc; ls Browse package documentation and examples cd /usr/local/share/examples On the web: www.freebsd.org Includes handbook, searchable mailing list archives System status Alt-F1 .. -

Inferring User Demographics and Social Strategies in Mobile Social Networks
Inferring User Demographics and Social Strategies in Mobile Social Networks Yuxiao Dongy, Yang Yangz, Jie Tangz, Yang Yangy, Nitesh V. Chawlay yDepartment of Computer Science and Engineering, University of Notre Dame yInterdisciplinary Center for Network Science and Applications, University of Notre Dame zDepartment of Computer Science and Technology, Tsinghua University [email protected], [email protected], [email protected], [email protected], [email protected] ABSTRACT 1. INTRODUCTION Demographics are widely used in marketing to characterize differ- From a recent report by the International Telecommunications ent types of customers. However, in practice, demographic infor- Union (ITU) at the 2013 Mobile World Congress, the number of mation such as age, gender, and location is usually unavailable due mobile-phone subscriptions reached 6.8 billion, corresponding to to privacy and other reasons. In this paper, we aim to harness the a global penetration of 96%. As of 2014, the number of mobile power of big data to automatically infer users’ demographics based subscriptions across the globe has exceeded the world population. on their daily mobile communication patterns. These mobile devices record huge amounts of user behavioral data, Our study is based on a real-world large mobile network of in particular users’ daily communications with others. This pro- more than 7,000,000 users and over 1,000,000,000 communication vides us with an unprecedented opportunity to study how people records (CALL and SMS). We discover several interesting social behave differently and form different groups. strategies that mobile users frequently use to maintain their so- Previous work on mobile social networks mainly focuses on cial connections. -

9 Shades of Lustre 04/2019 [email protected] Lustre Features Review
9 shades of Lustre 04/2019 [email protected] Lustre features review ►Metadata operations & optimizations • DNE • Lazy size on MDT ►File striping extensions • PFL • FLR use-case • DoM use case ►Lustre network aspects • Multi-Rail • Dynamic Peer Discovery • UDSP • LNet Health whamcloud.com Lustre metadata ls -l speed improvement performance scale out Simplifying network Choosing the correct configuration striping Increasing network Strengthening data bandwidth availability Small file performance improvement whamcloud.com Lustre metadata scale out ►Lustre initial design whamcloud.com Lustre metadata scale out ►DNE • aka Distributed Namespace Environment • phase 1 introduced with Lustre 2.4 (May 2013) whamcloud.com Lustre metadata scale out whamcloud.com Lustre metadata scale out ►DNE phase 1 benefits: • support up to 256 MDTs • additional MDTs on additional MDSes ►DNE phase 1 limitations: • remote dir assigned to a single MDT • only remote directory creation/unlink are allowed • no migration tool to move between MDTs (needs ‘mv’) • synchronous cross-MDT operations whamcloud.com Lustre metadata scale out ►DNE phase 2 • introduced with Lustre 2.8 (March 2016) • spread a single directory across multiple MDTs Þallow striped dir in addition to remote dir Þmuch more flexible than phase 1 Striped Directory Dir shard 0 Dir shard 1 Dir shard 2 Dir shard 3 fileA fileB fileC fileD whamcloud.com Lustre metadata scale out ►DNE phase 2 addresses phase 1 limitations: • rename and link ops supported • tool to migrate directories from one MDT to another -

Understanding and Troubleshooting Ports
UnderStanding and TroubleShooting Ports 1 This document is intended to assist users understand current state of the connection for any Port in the system. How does a system know to which port to address a communication? Many ports are defined by Internet standards as being used for a specific purpose or protocol. The list of ports can be viewed thru’ the below URL http://en.wikipedia.org/wiki/List_of_TCP_and_UDP_port_numbers Among the different Transport Protocol layers that use ports, users are likely to encounter User Datagram Protocol (UDP) and the Transmission Control Protocol (TCP) very often. There are a total of 65,535 TCP ports and a total of 65,535 different UDP ports. When a user informs that a communication is headed for a particular port (say port 53), then the next question that would usually follow is… if that is TCP port 53 or UDP port 53. For a port to be used to receive a network communication, the port must be associated with some process. The process acts as a listener, waiting for connections to be made requesting some service on its assigned port. In Windows, usually a Service is connected to a specific port (there might be exceptions as well). Using Netstat to analyze and understand Communication thru’ Ports The command “netstat” displays information about the network ports in use on the system. Netstat comes installed on all current releases of Windows systems. Run with no switches, netstat will simply display a list of active connections on the local system. A netstat command with no switches would get you an output similar to the below one (Few digits in IP and few characters in name are masked for security reasons). -

Oracle Single Client Access Name (SCAN)
An Oracle White Paper June 2013 Oracle Single Client Access Name (SCAN) Oracle Single Client Access Name (SCAN) Introduction ....................................................................................... 1 Network Requirements for Using SCAN ............................................ 2 Option 1 – Use the Corporate DNS ............................................... 2 Option 2 – Use the Oracle Grid Naming Service (GNS) ................. 4 Workaround if No DNS Server is Available at Installation Time ..... 4 SCAN Configuration with Oracle Grid Infrastructure 11g Release 2 .. 5 SCAN Configuration with Oracle Grid Infrastructure 12c Release 1 ... 6 Enabling Multiple-Subnet Support for SCAN .................................. 7 Oracle Database Configuration Using SCAN ................................... 10 Client Load Balancing using SCAN ............................................. 10 Multiple-Subnet Support and LISTENER_NETWORKS ............... 11 Version and Backward Compatibility ............................................... 12 Miscellaneous SCAN-related Configurations ................................... 13 Using SCAN with Multiple Ports on the Same Subnet.................. 13 Using SCAN in a MAA Environment not using GDS .................... 14 Using SCAN in a MAA Environment using GDS .......................... 14 Using SCAN with Oracle Connection Manager ............................ 15 Summary and Conclusion................................................................ 15 Oracle Single Client Access Name (SCAN) Introduction