Drug Metabolism in the Liver
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Detoxification Strategies for Zearalenone Using
microorganisms Review Detoxification Strategies for Zearalenone Using Microorganisms: A Review 1, 2, 1 1, Nan Wang y, Weiwei Wu y, Jiawen Pan and Miao Long * 1 Key Laboratory of Zoonosis of Liaoning Province, College of Animal Science & Veterinary Medicine, Shenyang Agricultural University, Shenyang 110866, China 2 Institute of Animal Science, Xinjiang Academy of Animal Sciences, Urumqi 830000, China * Correspondence: [email protected] or [email protected] These authors contributed equally to this work. y Received: 21 June 2019; Accepted: 19 July 2019; Published: 21 July 2019 Abstract: Zearalenone (ZEA) is a mycotoxin produced by Fusarium fungi that is commonly found in cereal crops. ZEA has an estrogen-like effect which affects the reproductive function of animals. It also damages the liver and kidneys and reduces immune function which leads to cytotoxicity and immunotoxicity. At present, the detoxification of mycotoxins is mainly accomplished using biological methods. Microbial-based methods involve zearalenone conversion or adsorption, but not all transformation products are nontoxic. In this paper, the non-pathogenic microorganisms which have been found to detoxify ZEA in recent years are summarized. Then, two mechanisms by which ZEA can be detoxified (adsorption and biotransformation) are discussed in more detail. The compounds produced by the subsequent degradation of ZEA and the heterogeneous expression of ZEA-degrading enzymes are also analyzed. The development trends in the use of probiotics as a ZEA detoxification strategy are also evaluated. The overall purpose of this paper is to provide a reliable reference strategy for the biological detoxification of ZEA. Keywords: zearalenone (ZEA); reproductive toxicity; cytotoxicity; immunotoxicity; biological detoxification; probiotics; ZEA biotransformation 1. -

Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development
Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development Gerlie Gieser, Ph.D. Office of Clinical Pharmacology, Div. IV Objectives • Outline the Phase 1 studies conducted to characterize the Clinical Pharmacology of a drug; describe important design elements of and the information gained from these studies. • List the Clinical Pharmacology characteristics of an Ideal Drug • Describe how the Clinical Pharmacology information from Phase 1 can help design Phase 2/3 trials • Discuss the timing of Clinical Pharmacology studies during drug development, and provide examples of how the information generated could impact the overall clinical development plan and product labeling. Phase 1 of Drug Development CLINICAL DEVELOPMENT RESEARCH PRE POST AND CLINICAL APPROVAL 1 DISCOVERY DEVELOPMENT 2 3 PHASE e e e s s s a a a h h h P P P Clinical Pharmacology Studies Initial IND (first in human) NDA/BLA SUBMISSION Phase 1 – studies designed mainly to investigate the safety/tolerability (if possible, identify MTD), pharmacokinetics and pharmacodynamics of an investigational drug in humans Clinical Pharmacology • Study of the Pharmacokinetics (PK) and Pharmacodynamics (PD) of the drug in humans – PK: what the body does to the drug (Absorption, Distribution, Metabolism, Excretion) – PD: what the drug does to the body • PK and PD profiles of the drug are influenced by physicochemical properties of the drug, product/formulation, administration route, patient’s intrinsic and extrinsic factors (e.g., organ dysfunction, diseases, concomitant medications, -

Medications to Treat Opioid Use Disorder Research Report
Research Report Revised Junio 2018 Medications to Treat Opioid Use Disorder Research Report Table of Contents Medications to Treat Opioid Use Disorder Research Report Overview How do medications to treat opioid use disorder work? How effective are medications to treat opioid use disorder? What are misconceptions about maintenance treatment? What is the treatment need versus the diversion risk for opioid use disorder treatment? What is the impact of medication for opioid use disorder treatment on HIV/HCV outcomes? How is opioid use disorder treated in the criminal justice system? Is medication to treat opioid use disorder available in the military? What treatment is available for pregnant mothers and their babies? How much does opioid treatment cost? Is naloxone accessible? References Page 1 Medications to Treat Opioid Use Disorder Research Report Discusses effective medications used to treat opioid use disorders: methadone, buprenorphine, and naltrexone. Overview An estimated 1.4 million people in the United States had a substance use disorder related to prescription opioids in 2019.1 However, only a fraction of people with prescription opioid use disorders receive tailored treatment (22 percent in 2019).1 Overdose deaths involving prescription opioids more than quadrupled from 1999 through 2016 followed by significant declines reported in both 2018 and 2019.2,3 Besides overdose, consequences of the opioid crisis include a rising incidence of infants born dependent on opioids because their mothers used these substances during pregnancy4,5 and increased spread of infectious diseases, including HIV and hepatitis C (HCV), as was seen in 2015 in southern Indiana.6 Effective prevention and treatment strategies exist for opioid misuse and use disorder but are highly underutilized across the United States. -

The Role of Detoxification in the Maintenance of Health Research
The Role of Detoxification in the have been reported as well (Table 1).2-11 Exposure to environmental toxicants can occur from air Maintenance of Health pollution, food supply, and drinking water, in addition to skin contact. For example, epidemiological studies have identified Research Review associations between symptoms of Parkinson’s disease and prolonged exposure to pesticides through farming or drinking TOXINS, TOXICANTS & TOXIC SUBSTANCES well water; proximity in residence to industrial plants, printing The word "toxin" itself does not describe a specific class of plants, or quarries; or chronic occupational exposure to 12 compounds, but rather something that can cause harm to the manganese, copper, or a combination of lead and iron. While body. More specifically, a toxin or toxic substance is a the mechanisms of these toxic exposures are not known, an chemical or mixture that may injure or present an individual’s ability to excrete toxins has been shown to be a 13,14 unreasonable risk of injury to the health of an exposed major factor in disease susceptibility. organism. The National Cancer Institute defines "toxin" as a poisonous compound made by bacteria, plants, or animals; it Table 1. Clinical Symptoms and Conditions Associated with defines “toxicant” as a poison made by humans or that is put Environmental Toxicity into the environment by human activities.1 Each toxic substance has a defined toxic dose or toxic concentration at Abnormal pregnancy outcomes which it produces its toxic effect. Atherosclerosis Broad mood swings Environmental pollutants (referred to as exogenous toxicants) Cancer present at variable levels in the air, drinking water, and food Chronic fatigue syndrome supply. -

Acute Liver Failure J G O’Grady
148 Postgrad Med J: first published as 10.1136/pgmj.2004.026005 on 4 March 2005. Downloaded from REVIEW Acute liver failure J G O’Grady ............................................................................................................................... Postgrad Med J 2005;81:148–154. doi: 10.1136/pgmj.2004.026005 Acute liver failure is a complex multisystemic illness that account for most cases, but a significant number of patients have no definable cause and are evolves quickly after a catastrophic insult to the liver classified as seronegative or of being of indeter- leading to the development of encephalopathy. The minate aetiology. Paracetamol is the commonest underlying aetiology and the pace of progression strongly cause in the UK and USA.2 Idiosyncratic reac- tions comprise another important group. influence the clinical course. The commonest causes are paracetamol, idiosyncratic drug reactions, hepatitis B, and Viral seronegative hepatitis. The optimal care is multidisciplinary ALF is an uncommon complication of viral and up to half of the cases receive liver transplants, with hepatitis, occurring in 0.2%–4% of cases depend- ing on the underlying aetiology.3 The risk is survival rates around 75%–90%. Artificial liver support lowest with hepatitis A, but it increases with the devices remain unproven in efficacy in acute liver failure. age at time of exposure. Hepatitis B can be associated with ALF through a number of ........................................................................... scenarios (table 2). The commonest are de novo infection and spontaneous surges in viral repli- cation, while the incidence of the delta virus cute liver failure (ALF) is a complex infection seems to be decreasing rapidly. multisystemic illness that evolves after a Vaccination should reduce the incidence of Acatastrophic insult to the liver manifesting hepatitis A and B, while antiviral drugs should in the development of a coagulopathy and ameliorate replication of hepatitis B. -

Help Prevent Relapse to Opioid Dependence After Opioid
For Opioid Dependence HELP REINFORCE YOUR RECOVERY Help prevent relapse to opioid dependence after opioid detoxification with a non-addictive, once-monthly treatment used with counseling.1,2 VIVITROL® is a prescription injectable medicine used to: Prevent relapse to opioid dependence after opioid detox. You must stop taking opioids or other opioid-containing medications before starting VIVITROL. To be effective, VIVITROL must be used with other alcohol or drug recovery programs, such as counseling. It is not known if VIVITROL is safe and effective in children. See important information about possible side effects with VIVITROL treatment throughout this brochure. Read the Brief Summary of Important Facts about VIVITROL on pages 5–6. This information does not take the place of talking with your healthcare provider. BRIEF SUMMARY OF IMPORTANT FACTS ABOUT VIVITROL ARE YOU OR YOUR LOVED ONE READY TO MOVE FORWARD? Opioid addiction is a chronic brain disease defined by an uncontrollable urge to seek and use opioids, like heroin or prescription pain medication. Because addiction changes the way the brain works, most patients need ongoing care in the form of counseling and medication.3 Discuss all benefits and risks of VIVITROL with your healthcare provider and whether VIVITROL may be right for you. Call your healthcare provider for medical advice about any side effects. PRESCRIBING See important information on possible side effects with VIVITROL treatment throughout this brochure. MEDICATION GUIDE Read the Brief Summary of Important Facts about VIVITROL by clicking the button in the top right-hand INFORMATION 2 corner. This information does not take the place of talking with your healthcare provider. -

13. Fundamentals of Toxicity and Detoxification.Pdf
FUNDAMENTALS OF TOXICITY AND DETOXIFICATION AND NUCLEAR MEDICINES BY Prof. Ramesh Chandra Department of Chemistry University of Delhi TOXICITY Toxicity The degree to which a substance (a toxin or poison) can harm humans or animals Acute toxicity involves harmful effects in an organism through a single or short-term exposure Subchronic toxicity is the ability of a toxic substance to cause effects for more than one year but less than the lifetime of the exposed organism. Chronic toxicity is the ability of a substance or mixture of substances to cause harmful effects over an extended period, usually upon repeated or continuous exposure, sometimes lasting for the entire life of the exposed organism. DETOXIFICATION NUCLEAR MEDICINES History 1946 first uses of nuclear medicine 1950s Widespread clinical use of nuclear medicine began 1960s measuring blood flow to the lungs and identifying cancer 1970s most organs of the body could be visualized with nuclear medicine procedures 1980s Radiopharmaceuticals, monoclonal antibodies, FDG 1990s PET 58 What is Nuclear Medicine? • Nuclear medicine is very unique, because it helps doctors view how your body is functioning. • This type of imaging takes very small amounts of radioactive pharmaceuticals and follows their path and progress through your body. • X-rays or CAT scans can show how something in your body looks, but Nuclear Medicine can show how your body actually works. What is Nuclear Medicine? (continued…) • Nuclear medicine is a type of molecular imaging where radioactive pharmaceuticals (often called “radiopharmaceuticals”) are used to evaluate the body’s functions and processes • This type of imaging can be used on all types of living things, but NMTCB is concerned with using this technology to help diagnose and treat human beings. -

Differential Metabolism of Alprazolam by Liver and Brain Cytochrome (P4503A) to Pharmacologically Active Metabolite
The Pharmacogenomics Journal (2002) 2, 243–258 2002 Nature Publishing Group All rights reserved 1470-269X/02 $25.00 www.nature.com/tpj ORIGINAL ARTICLE Differential metabolism of alprazolam by liver and brain cytochrome (P4503A) to pharmacologically active metabolite HV Pai1,2* ABSTRACT SC Upadhya1,2* Cytochrome P450 (P450) is a superfamily of enzymes which mediates metab- 1 olism of xenobiotics including drugs. Alprazolam, an anti-anxiety agent, is SJ Chinta * metabolized in rat and human liver by P4503A1 and P4503A4 respectively, SN Hegde1 to 4-hydroxy alprazolam (4-OHALP, pharmacologically less active) and ␣- V Ravindranath1,2 hydroxy alprazolam (␣-OHALP, pharmacologically more active). We exam- ined P450 mediated metabolism of alprazolam by rat and human brain 1Department of Neurochemistry, National microsomes and observed that the relative amount of ␣-OHALP formed in Institute of Mental Health & Neurosciences, brain was higher than liver. This biotransformation was mediated by a P450 Bangalore, India; 2National Brain Research Centre, ICGEB Campus, Aruna Asaf Ali Marg, isoform belonging to P4503A subfamily, which is constitutively expressed in New Delhi , India neuronal cells in rat and human brain. The formation of larger amounts of ␣-OHALP in neurons points to local modulation of pharmacological activity Correspondence: in brain, at the site of action of the anti-anxiety drug. Since hydroxy metab- V Ravindranath, National Brain Research olites of alprazolam are hydrophilic and not easily cleared through blood- Centre, ICGEB Campus, Aruna Asaf Ali ␣ Marg, New Delhi - 110 067, India CSF barrier, -OHALP would potentially have a longer half-life in brain. Tel: +91 124 630 8317 The Pharmacogenomics Journal (2002) 2, 243–258. -

Fact Sheet - Symptoms of Pancreatic Cancer
Fact Sheet - Symptoms of Pancreatic Cancer Diagnosis Pancreatic cancer is often difficult to diagnose, because the pancreas lies deep in the abdomen, behind the stomach, so tumors are not felt during a physical exam. Pancreatic cancer is often called the “silent” cancer because the tumor can grow for many years before it causes pressure, pain, or other signs of illness. When symptoms do appear, they can vary depending on the size of the tumor and where it is located on the pancreas. For these reasons, the symptoms of pancreatic cancer are seldom recognized until the cancer has progressed to an advanced stage and often spread to other areas of the body. General Symptoms Pain The first symptom of pancreatic cancer is often pain, because the tumors invade nerve clusters. Pain can be felt in the stomach area and/or in the back. The pain is generally worse after eating and when lying down, and is sometimes relieved by bending forward. Pain is more common in cancers of the body and tail of the pancreas. The abdomen may also be generally tender or painful if the liver, pancreas or gall bladder are inflamed or enlarged. It is important to keep in mind that there are many other causes of abdominal and back pain! Jaundice More than half of pancreatic cancer sufferers have jaundice, a yellowing of the skin and whites of the eyes. Jaundice is caused by a build-up bilirubin, a substance which is made in the liver and a component of bile. Bilirubin contains a lot of yellow pigment, and gives bile it’s color. -

Hepatitis B? HEPATITIS B Hepatitis B Is a Contagious Liver Disease That Results from Infection with the Hepatitis B Virus
What is Hepatitis B? HEPATITIS B Hepatitis B is a contagious liver disease that results from infection with the Hepatitis B virus. When first infected, a person can develop Are you at risk? an “acute” infection, which can range in severity from a very mild illness with few or no symptoms to a serious condition requiring hospitalization. Acute Hepatitis B refers to the first 6 months after someone is exposed to the Hepatitis B virus. Some people are able to fight the infection and clear the virus. For others, the infection remains and leads to a “chronic,” or lifelong, illness. Chronic Hepatitis B refers to the illness that occurs when the Hepatitis B virus remains in a person’s body. Over time, the infection can cause serious health problems. How is Hepatitis B spread? Hepatitis B is usually spread when blood, semen, or other body fluids from a person infected with the Hepatitis B virus enter the body of someone who is not infected. This can happen through having sex with an infected partner; sharing needles, syringes, or other injection drug equipment; or from direct contact with the blood or open sores of an infected person. Hepatitis B can also be passed from an infected mother to her baby at birth. Who should be tested for Hepatitis B? Approximately 1.2 million people in the United States and 350 million people worldwide have Hepatitis B. Testing for Hepatitis B is recommended for certain groups of people, including: Most are unaware of their infection. ■ People born in Asia, Africa, and other regions with moderate or high rates Is Hepatitis B common? of Hepatitis B (see map) Yes. -
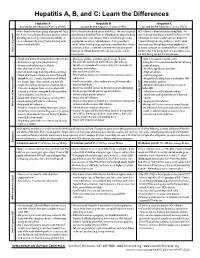
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Environmental Exposure / Detoxification
Environmental Exposure and Detoxification Gauge the Body’s Ability to Eliminate Toxins ■■ DNA Oxidative Damage ■■ Urine Porphyrins ■■ Glutathione, Erythrocytes ■■ DNA Methylation Profile ■■ Hepatic Detox Profile Science + Insight Environmental Exposure and Detoxification Environmental chemical exposure has never been more pervasive with thousands of chemicals in use around the world. Many chemicals are integrated into our food supply, the air we breathe and the water we drink. Every day, we ingest tiny amounts of these chemicals and our bodies cannot metabolize and clear all of them. Chemicals not metabolized are stored in the fat cells throughout our bodies, where they continue to accumulate. As these chemicals build up they alter our metabolism, cause enzyme dysfunction and nutritional deficiencies, create hormonal imbalances, damage brain chemistry and can cause cancer. Because the chemicals accumulate in different parts of the body—at different rates and in different combinations—there are many different chronic illnesses that can result. Doctor’s Data offers a spectrum of tests designed to evaluate the exposure to environmental toxins and markers of the body’s capacity for endogenous detoxification. The World Health Organization (WHO) estimates that about a quarter of the diseases facing mankind today occur due to prolonged exposure to environmental pollution. 2 DNA Oxidative Damage Oxidative stress has been associated Results are presented in a clear, easy-to-understand report. with many diseases, including bladder and prostate cancer, cystic fibrosis, atopic dermatitis, rheumatoid arthritis, and a wide range of neurological conditions, including Parkinson’s disease, Alzheimer’s disease and Huntington’s disease. It has also been correlated with the severity of diabetic retinopathy and neuropathy.