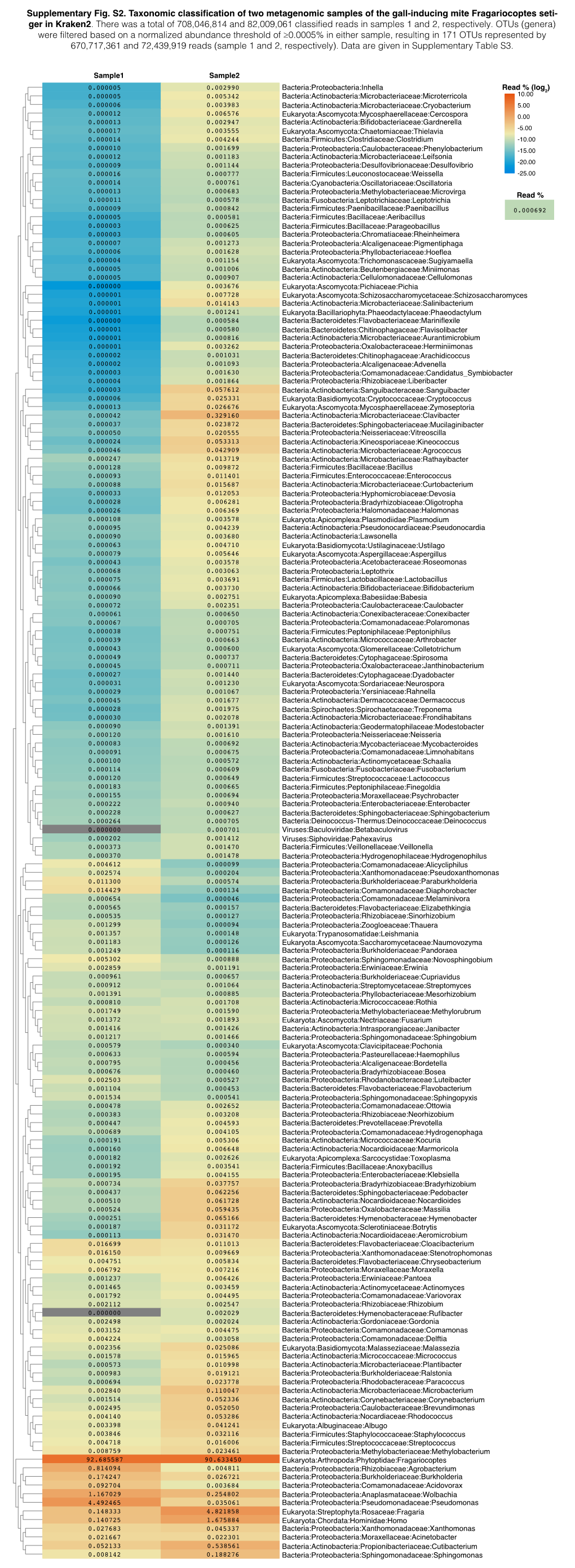
Supplementary Fig. S2. Taxonomic Classification of Two Metagenomic Samples of the Gall-Inducing Mite Fragariocoptes Seti- Ger in Kraken2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(Pentatomidae) DISSERTATION Presented
Genome Evolution During Development of Symbiosis in Extracellular Mutualists of Stink Bugs (Pentatomidae) DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Alejandro Otero-Bravo Graduate Program in Evolution, Ecology and Organismal Biology The Ohio State University 2020 Dissertation Committee: Zakee L. Sabree, Advisor Rachelle Adams Norman Johnson Laura Kubatko Copyrighted by Alejandro Otero-Bravo 2020 Abstract Nutritional symbioses between bacteria and insects are prevalent, diverse, and have allowed insects to expand their feeding strategies and niches. It has been well characterized that long-term insect-bacterial mutualisms cause genome reduction resulting in extremely small genomes, some even approaching sizes more similar to organelles than bacteria. While several symbioses have been described, each provides a limited view of a single or few stages of the process of reduction and the minority of these are of extracellular symbionts. This dissertation aims to address the knowledge gap in the genome evolution of extracellular insect symbionts using the stink bug – Pantoea system. Specifically, how do these symbionts genomes evolve and differ from their free- living or intracellular counterparts? In the introduction, we review the literature on extracellular symbionts of stink bugs and explore the characteristics of this system that make it valuable for the study of symbiosis. We find that stink bug symbiont genomes are very valuable for the study of genome evolution due not only to their biphasic lifestyle, but also to the degree of coevolution with their hosts. i In Chapter 1 we investigate one of the traits associated with genome reduction, high mutation rates, for Candidatus ‘Pantoea carbekii’ the symbiont of the economically important pest insect Halyomorpha halys, the brown marmorated stink bug, and evaluate its potential for elucidating host distribution, an analysis which has been successfully used with other intracellular symbionts. -

Pfc5813.Pdf (9.887Mb)
UNIVERSIDAD POLITÉCNICA DE CARTAGENA ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA DEPARTAMENTO DE PRODUCCIÓN VEGETAL INGENIERO AGRÓNOMO PROYECTO FIN DE CARRERA: “AISLAMIENTO E IDENTIFICACIÓN DE LOS RIZOBIOS ASOCIADOS A LOS NÓDULOS DE ASTRAGALUS NITIDIFLORUS”. Realizado por: Noelia Real Giménez Dirigido por: María José Vicente Colomer Francisco José Segura Carreras Cartagena, Julio de 2014. ÍNDICE GENERAL 1. Introducción…………………………………………………….…………………………………………………1 1.1. Astragalus nitidiflorus………………………………..…………………………………………………2 1.1.1. Encuadre taxonómico……………………………….…..………………………………………………2 1.1.2. El origen de Astragalus nitidiflorus………………………………………………………………..4 1.1.3. Descripción de la especie………..…………………………………………………………………….5 1.1.4. Biología…………………………………………………………………………………………………………7 1.1.4.1. Ciclo vegetativo………………….……………………………………………………………………7 1.1.4.2. Fenología de la floración……………………………………………………………………….9 1.1.4.3. Sistema de reproducción……………………………………………………………………….10 1.1.4.4. Dispersión de los frutos…………………………………….…………………………………..11 1.1.4.5. Nodulación con Rhizobium…………………………………………………………………….12 1.1.4.6. Diversidad genética……………………………………………………………………………....13 1.1.5. Ecología………………………………………………………………………………………………..…….14 1.1.6. Corología y tamaño poblacional……………………………………………………..…………..15 1.1.7. Protección…………………………………………………………………………………………………..18 1.1.8. Amenazas……………………………………………………………………………………………………19 1.1.8.1. Factores bióticos…………………………………………………………………………………..19 1.1.8.2. Factores abióticos………………………………………………………………………………….20 1.1.8.3. Factores antrópicos………………..…………………………………………………………….21 -

S13568-021-01252-2.Pdf
Chen et al. AMB Expr (2021) 11:93 https://doi.org/10.1186/s13568-021-01252-2 ORIGINAL ARTICLE Open Access Habitat environmental factors infuence intestinal microbial diversity of the short-faced moles (Scaptochirus moschata) Lei Chen*, Di Xu, Jing Zhu, Shen Wang, Mi Liu, Mengyao Sun, Geyang Wang, Lingyu Song, Xiaoyu Liu and Tianyu Xie Abstract The short-faced moles (Scaptochirus moschata) are unique Chinese mammal that live in burrows for life. They have complex ecological adaptation mechanisms to adapt to perennial underground life. Intestinal microbes play an important role in the ecological adaptation of wild animals. The gut microbiota diversity and its function in short- faced moles’ ecological adaptation is a scientifc issue worth exploring. In this study, the Illumina HiSeq sequencing platform was used to sequence the V3-V4 hypervariable regions of the 16S rRNA genes of 22 short-faced moles’ intes- tinal samples to study the composition and functional structure of their intestinal microbiota. The results showed that in the short-faced moles’ intestine, there are four main phyla, Firmicutes, Proteobacteria, Actinobacteria and Bacteroidete. At the family level, Peptostreptococcaceae and Enterobacteriaceae have the highest abundance. At the genus level, Romboutsia is the genus with the highest microbial abundance. According to the KEGG database, the main func- tions of short-faced mole gut microbes are metabolism, genetic information processing, environmental information processing, and cellular processes. The function of short-faced mole intestinal microbiota is suitable for its long-term burrowing life. No gender diference is found in the composition and function of the short-faced mole intestinal microbiota. -

First Report of Pathogenic Bacterium Kalamiella Piersonii Isolated
pathogens Article First Report of Pathogenic Bacterium Kalamiella piersonii Isolated from Urine of a Kidney Stone Patient: Draft Genome and Evidence for Role in Struvite Crystallization 1, 1,2, 1, Punchappady Devasya Rekha *, Asif Hameed y, Muhammed A. P. Manzoor y , 1, 1 1 1, 1, Mangesh V. Suryavanshi y , Sudeep D. Ghate , A. B. Arun , Sneha S. Rao y, Athmika y, Sukesh Kumar Bajire 1, M. Mujeeburahiman 3 and C.-C. Young 2 1 Yenepoya Research Centre, Yenepoya Deemed to be University, Mangalore 575018, India; [email protected] (A.H.); [email protected] (M.A.P.M.); [email protected] (M.V.S.); [email protected] (S.D.G.); [email protected] (A.B.A.); [email protected] (S.S.R.); [email protected] (A.); [email protected] (S.K.B.) 2 Department of Soil and Environmental Sciences, College of Agriculture and Natural Resources, National Chung Hsing University, Taichung 40227, Taiwan; [email protected] 3 Department of Urology, Yenepoya Medical College and Hospital, Yenepoya Deemed to be University, Mangalore 575018, India; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 24 July 2020; Accepted: 17 August 2020; Published: 29 August 2020 Abstract: Uropathogenic bacteria are widely distributed in the environment and urinary tract infection is implicated in kidney stone disease. Here, we report on a urease negative bacterium Kalamiella piersonii (strain YU22) isolated from the urine of a struvite stone (MgNH PO 6H O) 4 4· 2 patient. The closest species, K. piersonii IIIF1SW-P2T was reported from International Space Station samples. -

The Subway Microbiome: Seasonal Dynamics and Direct Comparison Of
Gohli et al. Microbiome (2019) 7:160 https://doi.org/10.1186/s40168-019-0772-9 RESEARCH Open Access The subway microbiome: seasonal dynamics and direct comparison of air and surface bacterial communities Jostein Gohli1* , Kari Oline Bøifot1,2, Line Victoria Moen1, Paulina Pastuszek3, Gunnar Skogan1, Klas I. Udekwu4 and Marius Dybwad1,2 Abstract Background: Mass transit environments, such as subways, are uniquely important for transmission of microbes among humans and built environments, and for their ability to spread pathogens and impact large numbers of people. In order to gain a deeper understanding of microbiome dynamics in subways, we must identify variables that affect microbial composition and those microorganisms that are unique to specific habitats. Methods: We performed high-throughput 16S rRNA gene sequencing of air and surface samples from 16 subway stations in Oslo, Norway, across all four seasons. Distinguishing features across seasons and between air and surface were identified using random forest classification analyses, followed by in-depth diversity analyses. Results: There were significant differences between the air and surface bacterial communities, and across seasons. Highly abundant groups were generally ubiquitous; however, a large number of taxa with low prevalence and abundance were exclusively present in only one sample matrix or one season. Among the highly abundant families and genera, we found that some were uniquely so in air samples. In surface samples, all highly abundant groups were also well represented in air samples. This is congruent with a pattern observed for the entire dataset, namely that air samples had significantly higher within-sample diversity. We also observed a seasonal pattern: diversity was higher during spring and summer. -

Isolation and Identification of Microvirga Thermotolerans HR1, A
microorganisms Article Isolation and Identification of Microvirga thermotolerans HR1, a Novel Thermo-Tolerant Bacterium, and Comparative Genomics among Microvirga Species Jiang Li 1,2, Ruyu Gao 2, Yun Chen 2, Dong Xue 2, Jiahui Han 2, Jin Wang 1,2, Qilin Dai 1, Min Lin 2, Xiubin Ke 2,* and Wei Zhang 2,* 1 School of Life Science and Engineering, Southwest University of Science and Technology, Mianyang 621010, Sichuan, China; [email protected] (J.L.); [email protected] (J.W.); [email protected] (Q.D.) 2 Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing 100081, China; [email protected] (R.G.); [email protected] (Y.C.); [email protected] (D.X.); [email protected] (J.H.); [email protected] (M.L.) * Correspondence: [email protected] (X.K.); [email protected] (W.Z.) Received: 27 November 2019; Accepted: 9 January 2020; Published: 10 January 2020 Abstract: Members of the Microvirga genus are metabolically versatile and widely distributed in Nature. However, knowledge of the bacteria that belong to this genus is currently limited to biochemical characteristics. Herein, a novel thermo-tolerant bacterium named Microvirga thermotolerans HR1 was isolated and identified. Based on the 16S rRNA gene sequence analysis, the strain HR1 belonged to the genus Microvirga and was highly similar to Microvirga sp. 17 mud 1-3. The strain could grow at temperatures ranging from 15 to 50 ◦C with a growth optimum at 40 ◦C. It exhibited tolerance to pH range of 6.0–8.0 and salt concentrations up to 0.5% (w/v). It contained ubiquinone 10 as the predominant quinone and added group 8 as the main fatty acids. -

Changes in Multi-Level Biodiversity and Soil Features in a Burned Beech Forest in the Southern Italian Coastal Mountain
Article Changes in Multi-Level Biodiversity and Soil Features in a Burned Beech Forest in the Southern Italian Coastal Mountain Adriano Stinca 1,* , Maria Ravo 2, Rossana Marzaioli 1,*, Giovanna Marchese 2, Angela Cordella 2, Flora A. Rutigliano 1 and Assunta Esposito 1 1 Department of Environmental Biological and Pharmaceutical Sciences and Technologies, University of Campania Luigi Vanvitelli, Via Vivaldi 43, 81100 Caserta, Italy; fl[email protected] (F.A.R.); [email protected] (A.E.) 2 Genomix4Life S.r.l., Via Allende, 84081 Baronissi (Salerno), Italy; [email protected] (M.R.); [email protected] (G.M.); [email protected] (A.C.) * Correspondence: [email protected] (A.S.); [email protected] (R.M.) Received: 1 August 2020; Accepted: 8 September 2020; Published: 11 September 2020 Abstract: In the context of global warming and increasing wildfire occurrence, this study aims to examine, for the first time, the changes in multi-level biodiversity and key soil features related to soil functioning in a burned Mediterranean beech forest. Two years after the 2017 wildfire, changes between burned and unburned plots of beech forest were analyzed for plant communities (vascular plant and cover, bryophytes diversity, structural, chorological, and ecological variables) and soil features (main chemical properties, microbial biomass and activity, bacterial community composition, and diversity), through a synchronic study. Fire-induced changes in the micro-environmental conditions triggered a secondary succession process with colonization by many native pioneer plant species. Indeed, higher frequency (e.g., Scrophularia vernalis L., Rubus hirtus Waldst. and Kit. group, and Funaria hygrometrica Hedw.) or coverage (e.g., Verbascum thapsus L. -

Diversion and Phylogenetic Relatedness of Filterable Bacteria from Norwegian Tap and Bottled Waters
Diversion and phylogenetic relatedness of filterable bacteria from Norwegian tap and bottled waters Short title: Filterable bacteria in Norwegian tap and bottled waters Colin Charnock*, Ralf Xue Hagen, Theresa Ngoc-Thu Nguyen, Linh Thuy Vo Department of Life Sciences and Health, Oslo Metropolitan University, NO-0130, Oslo, Norway *Corresponding author. E-mail address: [email protected]. ABSTRACT Numerous articles have documented the existence of filterable bacteria. Where filtration is the chosen method of sterilization for medicinal or media components, these bacteria will by definition render products non-sterile. They may further represent a health hazard to the end user. A wide-range of bacterial genera were found in bottled and tap water filtrates from 0.2 µm filters, including genera housing opportunistic pathogens (e.g. Methylobacterium) and endospore formers (Paenibacillus). Two municipal tap water isolates were only distantly related to named species. One of these grew on agar, and could potentially provide hitherto unharvested useful biological products. The other grew only in water, and failed to produce colonies on media targeting either heterotrophs or autotrophs. The present study is one of very few looking at filterable bacteria in bottled waters intended for human consumption and the first identifying the filterable portion. It extends the range of known habitats of filterable bacteria and provides data on two new or novel species. Key words │bottled water, filterable bacteria, new species ©IWA Publishing 2019. The definitive peer-reviewed and edited version of this article is published in J Water Health (2019) 17 (2): 295-307 https://doi.org/10.2166/wh.2019.284 and is available at www.iwapublishing.com. -

Stress-Tolerance and Taxonomy of Culturable Bacterial Communities Isolated from a Central Mojave Desert Soil Sample
geosciences Article Stress-Tolerance and Taxonomy of Culturable Bacterial Communities Isolated from a Central Mojave Desert Soil Sample Andrey A. Belov 1,*, Vladimir S. Cheptsov 1,2 , Elena A. Vorobyova 1,2, Natalia A. Manucharova 1 and Zakhar S. Ezhelev 1 1 Soil Science Faculty, Lomonosov Moscow State University, Moscow 119991, Russia; [email protected] (V.S.C.); [email protected] (E.A.V.); [email protected] (N.A.M.); [email protected] (Z.S.E.) 2 Space Research Institute, Russian Academy of Sciences, Moscow 119991, Russia * Correspondence: [email protected]; Tel.: +7-917-584-44-07 Received: 28 February 2019; Accepted: 8 April 2019; Published: 10 April 2019 Abstract: The arid Mojave Desert is one of the most significant terrestrial analogue objects for astrobiological research due to its genesis, mineralogy, and climate. However, the knowledge of culturable bacterial communities found in this extreme ecotope’s soil is yet insufficient. Therefore, our research has been aimed to fulfil this lack of knowledge and improve the understanding of functioning of edaphic bacterial communities of the Central Mojave Desert soil. We characterized aerobic heterotrophic soil bacterial communities of the central region of the Mojave Desert. A high total number of prokaryotic cells and a high proportion of culturable forms in the soil studied were observed. Prevalence of Actinobacteria, Proteobacteria, and Firmicutes was discovered. The dominance of pigmented strains in culturable communities and high proportion of thermotolerant and pH-tolerant bacteria were detected. Resistance to a number of salts, including the ones found in Martian regolith, as well as antibiotic resistance, were also estimated. -

Table S4. Phylogenetic Distribution of Bacterial and Archaea Genomes in Groups A, B, C, D, and X
Table S4. Phylogenetic distribution of bacterial and archaea genomes in groups A, B, C, D, and X. Group A a: Total number of genomes in the taxon b: Number of group A genomes in the taxon c: Percentage of group A genomes in the taxon a b c cellular organisms 5007 2974 59.4 |__ Bacteria 4769 2935 61.5 | |__ Proteobacteria 1854 1570 84.7 | | |__ Gammaproteobacteria 711 631 88.7 | | | |__ Enterobacterales 112 97 86.6 | | | | |__ Enterobacteriaceae 41 32 78.0 | | | | | |__ unclassified Enterobacteriaceae 13 7 53.8 | | | | |__ Erwiniaceae 30 28 93.3 | | | | | |__ Erwinia 10 10 100.0 | | | | | |__ Buchnera 8 8 100.0 | | | | | | |__ Buchnera aphidicola 8 8 100.0 | | | | | |__ Pantoea 8 8 100.0 | | | | |__ Yersiniaceae 14 14 100.0 | | | | | |__ Serratia 8 8 100.0 | | | | |__ Morganellaceae 13 10 76.9 | | | | |__ Pectobacteriaceae 8 8 100.0 | | | |__ Alteromonadales 94 94 100.0 | | | | |__ Alteromonadaceae 34 34 100.0 | | | | | |__ Marinobacter 12 12 100.0 | | | | |__ Shewanellaceae 17 17 100.0 | | | | | |__ Shewanella 17 17 100.0 | | | | |__ Pseudoalteromonadaceae 16 16 100.0 | | | | | |__ Pseudoalteromonas 15 15 100.0 | | | | |__ Idiomarinaceae 9 9 100.0 | | | | | |__ Idiomarina 9 9 100.0 | | | | |__ Colwelliaceae 6 6 100.0 | | | |__ Pseudomonadales 81 81 100.0 | | | | |__ Moraxellaceae 41 41 100.0 | | | | | |__ Acinetobacter 25 25 100.0 | | | | | |__ Psychrobacter 8 8 100.0 | | | | | |__ Moraxella 6 6 100.0 | | | | |__ Pseudomonadaceae 40 40 100.0 | | | | | |__ Pseudomonas 38 38 100.0 | | | |__ Oceanospirillales 73 72 98.6 | | | | |__ Oceanospirillaceae -

16S Rrna Metagenomic Analysis of the Bacterial Community Associated with Turf Grass Seeds from Low Moisture and High Moisture Climates
16S rRNA metagenomic analysis of the bacterial community associated with turf grass seeds from low moisture and high moisture climates Qiang Chen, William A. Meyer, Qiuwei Zhang and James F. White Department of Plant Biology, Rutgers, The State University of New Jersey, New Brunswick, NJ, USA ABSTRACT Turfgrass investigators have observed that plantings of grass seeds produced in moist climates produce seedling stands that show greater stand evenness with reduced disease compared to those grown from seeds produced in dry climates. Grass seeds carry microbes on their surfaces that become endophytic in seedlings and promote seedling growth. We hypothesize that incomplete development of the microbiome associated with the surface of seeds produced in dry climates reduces the performance of seeds. Little is known about the influence of moisture on the structure of this microbial community. We conducted metagenomic analysis of the bacterial communities associated with seeds of three turf species (Festuca rubra, Lolium arundinacea, and Lolium perenne) from low moisture (LM) and high moisture (HM) climates. The bacterial communities were characterized by Illumina high-throughput sequencing of 16S rRNA V3–V4 regions. We performed seed germination tests and analyzed the correlations between the abundance of different bacterial groups and seed germination at different taxonomy ranks. Climate appeared to structure the bacterial communities associated with seeds. LM seeds vectored mainly Proteobacteria (89%). HM seeds vectored a denser and more diverse bacterial community that included Proteobacteria (50%) and Bacteroides (39%). Submitted 29 August 2019 At the genus level, Pedobacter (20%), Sphingomonas (13%), Massilia (12%), Pantoea Accepted 16 December 2019 (12%) and Pseudomonas (11%) were the major genera in the bacterial communities Published 10 January 2020 regardless of climate conditions. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341