Development of Bacillus Subtilis Spores and Cells for Surface Display of Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Peptide Tag Systems That Spontaneously Form An
(19) TZZ ¥_T (11) EP 2 534 484 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: G01N 33/531 (2006.01) C07K 14/315 (2006.01) 19.11.2014 Bulletin 2014/47 C12N 15/00 (2006.01) (21) Application number: 11706621.7 (86) International application number: PCT/GB2011/000188 (22) Date of filing: 11.02.2011 (87) International publication number: WO 2011/098772 (18.08.2011 Gazette 2011/33) (54) PEPTIDE TAG SYSTEMS THAT SPONTANEOUSLY FORM AN IRREVERSIBLE LINK TO PROTEIN PARTNERS VIA ISOPEPTIDE BONDS PEPTIDMARKIERUNGSSYSTEME MIT SPONTANER BILDUNG EINER IRREVERSIBLEN VERBINDUNG ZU PROTEINPARTNERN ÜBER ISOPEPTIDBINDUNGEN SYSTÈMES DE MARQUAGE PEPTIDIQUE QUI FORMENT SPONTANÉMENT UNE LIAISON IRRÉVERSIBLE AVEC DES PARTENAIRES PROTÉIQUES PAR L’INTERMÉDIAIRE DE LIAISONS ISOPEPTIDIQUES (84) Designated Contracting States: • TOMINAGA J ET AL: "Design of a specific peptide AL AT BE BG CH CY CZ DE DK EE ES FI FR GB tagthat affords covalent and site- specificenzyme GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO immobilization catalyzed by microbial PL PT RO RS SE SI SK SM TR transglutaminase", BIOMACROMOLECULES JULY/AUGUST 2005 AMERICAN CHEMICAL (30) Priority: 11.02.2010 GB 201002362 SOCIETY US, vol. 6, no. 4, July 2005 (2005-07), pages 2299-2304, XP002631438, DOI: DOI: (43) Date of publication of application: 10.1021/BM050193O 19.12.2012 Bulletin 2012/51 • DONG RUI-PING ET AL: "Characterization of T cell epitopes restricted by HLA-DP9 in (73) Proprietor: Isis Innovation Limited streptococcal M12 protein", JOURNAL OF Summertown, Oxford OX2 7SQ (GB) IMMUNOLOGY, vol. -

Improving Protein Therapeutics Through Quantitative Molecular Engineering Approaches and a Cell-Based Oral Delivery Platform
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2013 Improving Protein Therapeutics Through Quantitative Molecular Engineering Approaches and A Cell-Based Oral Delivery Platform Ting Wun Ng University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biomedical Commons Recommended Citation Ng, Ting Wun, "Improving Protein Therapeutics Through Quantitative Molecular Engineering Approaches and A Cell-Based Oral Delivery Platform" (2013). Publicly Accessible Penn Dissertations. 784. https://repository.upenn.edu/edissertations/784 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/784 For more information, please contact [email protected]. Improving Protein Therapeutics Through Quantitative Molecular Engineering Approaches and A Cell-Based Oral Delivery Platform Abstract Proteins, with their ability to perform a variety of highly specific biological functions, have emerged as an important class of therapeutics. However, to fully harness their therapeutic potential, proteins often need to be optimized by molecular engineering; therapeutic efficacy can be improved by modulating protein properties such as binding affinity/specificity, half-life, bioavailability, and immunogenicity. In this work, we first present an introductory example in which a mechanistic mathematical model was used to improve target selection for directed evolution of an aglycosylated Fc domain of an antibody to enhance -

A Bacterial Display System for Effective Selection of Protein-Biotin Ligase Bira Variants with Novel Peptide Specificity
bioRxiv preprint doi: https://doi.org/10.1101/367730; this version posted July 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A bacterial display system for effective selection of protein-biotin ligase BirA variants with novel peptide specificity Jeff Granhøj, Henrik Dimke and Per Svenningsen* Department of Molecular Medicine, University of Southern Denmark, Odense, Denmark * Corresponding author: Dr. Per Svenningsen, Institute of Molecular Medicine, University of Southern Denmark, J.B. Winsloews vej 21.3, DK-5000 Odense C, Denmark. Running title: Bacterial display of enzymatic peptide biotinylation 1 bioRxiv preprint doi: https://doi.org/10.1101/367730; this version posted July 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Biotinylation creates a sensitive and specific tag for purification and detection of target proteins. The E. coli protein-biotin ligase BirA biotinylates a lysine within a synthetic biotin acceptor peptide (AP) and allow for specific tagging of proteins fused to the AP. The approach is not applicable to unmodified proteins, and we sought to develop an effective selection system that could form the basis for directed evolution of novel BirA variants with specificity towards unmodified proteins. The system was based on bacterial display of a target peptide sequence, which could be biotinylated by cytosolic BirA variants before being displayed on the surface. In a model selection, the bacterial display system accomplished >1.000.000 enrichment in a single selection step. -

Supporting Information for a Chemically Synthesized Capture
Supporting Information for A Chemically Synthesized Capture Agent Enables the Selective, Sensitive, and Robust Electrochemical Detection of Anthrax Protective Antigen Blake Farrow 1,2,§ , Sung A Hong 3,§ , Errika C. Romero 2, Bert Lai 4, Matthew B. Coppock 5, Kaycie M. Deyle 2, Amethist S. Finch 5, Dimitra N. Stratis-Cullum 5, Heather D. Agnew 4, Sung Yang 3,* and James R. Heath 2,* 1. Department of Applied Physics and Materials Science, and 2. Division of Chemistry and Chemical Engineering California Institute of Technology 1200 East California Boulevard, Pasadena, California 91125 3. Department of Medical System Engineering Gwangju Institute of Science and Technology Gwangju, 500712, Republic of Korea 4. Indi Molecular 6162 Bristol Parkway, Culver City, California 90230 5. Biotechnology Branch, Sensors & Electronic Devices Directorate U.S. Army Research Laboratory 2800 Powder Mill Rd. Adelphi, MD 20783 § These authors contributed equally to this work. 1 OH (e) NH 2 O NH N NH HN HN O O NH H 2 N O N O H2N NH H O O O O N O O O HN H NH NH N O NH O HN O H O O N O H N O NH 2 O O H O N O NH H N NH O O N O O O O O H2N O C O HN NH H H N O S NH H Figure S1: Screening protocol and anchor ligand. (a-d), Detailed screening steps for identifying biligand capture agent candidates. The details of this protocol are provided in the Supplementary Methods. (b) Structure of anchor ligand used in the biligand screen. -

Chapter 1 Ribosome Display
Chapter 1 Ribosome Display: A Perspective Andreas Plückthun Abstract Ribosome display is an in vitro evolution technology for proteins. It is based on in vitro translation, but prevents the newly synthesized protein and the mRNA encoding it from leaving the ribosome . It thereby couples phenotype and genotype. Since no cells need to be transformed, very large libraries can be used directly in selections, and the in vitro amplifi cation provides a very convenient integration of random mutagenesis that can be incorporated into the procedure. This review highlights concepts, mechanisms, and different variations of ribosome display and compares it to related methods. Applications of ribosome display are summarized, e.g., the directed evolution of proteins for higher binding affi nity, for higher stability or other improved biophysical parameters and enzymatic properties. Ribosome display has developed into a robust technology used in academia and industry alike, and it has made the cell-free Darwinian evolution of proteins over multiple generations a reality. Key words: Directed evolution , Cell-free translation, Ribosome display , Protein engineering, Antibody engineering , DARPins , Designed ankyrin repeat proteins , Affi nity maturation 1. Introduction: Ribosome Display in Context All technologies of molecular evolution must couple phenotype and genotype. There are two fundamental possibilities for achieving this. The fi rst one is compartmentalization. Nature’s compartments are cells: they secure that the superior phenotype expressed by one cell’s mutant genotype can be replicated, without the gene products from the wild type interfering. All selections based on microbial phenotypes use this principle. The second possibility is a direct physical coupling of genetic material to the protein product. -

Antibody Display Technologies: Selecting the Cream of the Crop
Biol. Chem. 2021; aop Review Bernhard Valldorf, Steffen C. Hinz, Giulio Russo, Lukas Pekar, Laura Mohr, Janina Klemm, Achim Doerner, Simon Krah, Michael Hust and Stefan Zielonka* Antibody display technologies: selecting the cream of the crop https://doi.org/10.1515/hsz-2020-0377 other technologies gained a certain level of maturity, most Received November 25, 2020; accepted March 5, 2021; strikingly mammalian display. In this review, we delineate published online March 23, 2021 the most important selection systems with respect to antibody generation with an emphasis on recent Abstract: Antibody display technologies enable the suc- developments. cessful isolation of antigen-specific antibodies with ther- apeutic potential. The key feature that facilitates the Keywords: antibody display; antibody engineering; anti- selection of an antibody with prescribed properties is the body screening; antibody selection; genotype phenotype coupling of the protein variant to its genetic information coupling. and is referred to as genotype phenotype coupling. There are several different platform technologies based on pro- karyotic organisms as well as strategies employing higher Introduction eukaryotes. Among those, phage display is the most established system with more than a dozen of therapeutic Monoclonal antibodies (mAbs) have proven to be remark- antibodies approved for therapy that have been discovered ably versatile therapeutics for disease treatment including or engineered using this approach. In recent years several cancer and inflammatory disorders (Chan and Carter 2010; Scott et al. 2012; Yasunaga, 2020). This is exemplified by Bernhard Valldorf, Steffen C. Hinz, Giulio Russo, and Lukas Pekar the fact that as of November 2019, around 80 mAbs have contributed equally to this work. -

Understanding Ubiquitin Recognition and Generating Affinity Reagents
Ubiquitin Engineering: Understanding Ubiquitin Recognition and Generating Affinity Reagents by Isabel Leung A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Isabel Leung 2017 Abstract Ubiquitin Engineering: Understanding Ubiquitin Recognition and Generating Affinity Reagents Isabel Leung Doctor of Philosophy Department of Molecular Genetics University of Toronto 2016 Protein-protein interactions are necessary for virtually all biological processes. There have been tremendous efforts to document the diversity of molecular recognition, and to understand how molecular recognition occurs. The understanding of molecular interaction has also served as the foundation for designing novel protein interactions for use in therapeutics, diagnostics and basic sciences. An attractive system for studying protein-protein interactions is the ubiquitin (Ub) system. Ub is a protein modifier that is combinatorially ligated onto substrate proteins to influence substrate turnover and function. Ub uses a common surface to interact with more than 1000 proteins and plays pivotal roles in cell physiology. Despite the substantial structural information on Ub mediated interactions, there is no clear understanding of how individual Ub residues contribute to Ub’s broad scope of interactions. To address this question, I used affinity enhanced Ub variants (Ubvs) as proxies of native Ub in saturation scanning. Using saturation scanning, I studied the interactions between Ubvs and two Ub specific proteases (USP), USP2 and USP21, and elucidated a common functional epitope that is critical for USP recognition. The functional epitope recognizes USP residues that are conserved among the human USP family, suggesting it may make functional contributions in many other USP interactions. -

Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands
International Journal of Molecular Sciences Article Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands John Bowen 1, John Schneible 1 , Kaitlyn Bacon 1, Collin Labar 2, Stefano Menegatti 1,3,* and Balaji M. Rao 1,3,* 1 Department of Chemical and Biomolecular Engineering, North Carolina State University, 911 Partners Way, Raleigh, NC 27695, USA; [email protected] (J.B.); [email protected] (J.S.); [email protected] (K.B.) 2 Department of Molecular and Structural Biochemistry, North Carolina State University, Raleigh, NC 27695, USA; [email protected] 3 Biomanufacturing Training and Education Center (BTEC), North Carolina State University, 850 Oval Dr, Raleigh, NC 27606, USA * Correspondence: [email protected] (S.M.); [email protected] (B.M.R.) Abstract: We present the construction and screening of yeast display libraries of post-translationally modified peptides wherein site-selective enzymatic treatment of linear peptides is achieved using bacterial transglutaminase. To this end, we developed two alternative routes, namely (i) yeast display of linear peptides followed by treatment with recombinant transglutaminase in solution; or (ii) intracellular co-expression of linear peptides and transglutaminase to achieve peptide modification in the endoplasmic reticulum prior to yeast surface display. The efficiency of peptide modification was evaluated via orthogonal detection of epitope tags integrated in the yeast-displayed peptides by flow cytometry, and via comparative cleavage of putative cyclic vs. linear peptides by tobacco etch virus (TEV) protease. Subsequently, yeast display libraries of transglutaminase-treated peptides were screened to isolate binders to the N-terminal region of the Yes-Associated Protein (YAP) and its WW domains using magnetic selection and fluorescence activated cell sorting (FACS). -

Deep Mutational Analysis Reveals Functional Trade-Offs in the Sequences of EGFR Autophosphorylation Sites
Deep mutational analysis reveals functional trade-offs in the sequences of EGFR autophosphorylation sites Aaron J. Cantora,b,c, Neel H. Shaha,b,c, and John Kuriyana,b,c,d,e,1 aDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720; bDepartment of Chemistry, University of California, Berkeley, CA 94720; cCalifornia Institute for Quantitative Biosciences, University of California, Berkeley, CA 94720; dHoward Hughes Medical Institute, University of California, Berkeley, CA 94720; and eMolecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720 Edited by John D. Scott, HHMI and Department of Pharmacology, University of Washington School of Medicine, Seattle, WA, and accepted by Editorial Board Member Brenda A. Schulman June 18, 2018 (received for review March 1, 2018) Upon activation, the epidermal growth factor receptor (EGFR) substrate sequences over others, with specificity being determined phosphorylates tyrosine residues in its cytoplasmic tail, which by the pattern of amino acid residues directly adjacent to triggers the binding of Src homology 2 (SH2) and phosphotyrosine- the tyrosine (12, 13). binding (PTB) domains and initiates downstream signaling. The For EGFR-family members, the high local concentration of sequences flanking the tyrosine residues (referred to as “phospho- tail phosphosites with respect to the kinase domains may allow sites”) must be compatible with phosphorylation by the EGFR kinase these sites to be phosphorylated without much regard for se- domain and the recruitment of adapter proteins, while minimizing quence. This made us wonder about the extent to which each phosphorylation that would reduce the fidelity of signal transmis- phosphosite in the cytoplasmic tails of EGFR-family members is sion. -
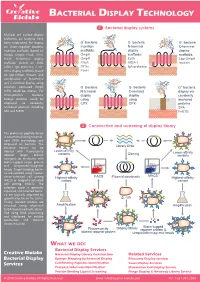
Bacterial Display Technology
BACTERIAL DISPLAY TECHNOLOGY 1 Bacterial display systems Multiple cell surface display platforms on bacteria have - - - been established. For display G bacteria G bacteria G bacteria on Gram-negative bacteria, Insertion N-terminal C-terminal insertion scaffolds (based on scaffolds: N display display C OmpA, OmpX, FhuA, FliTrx, OmpA scaffolds: scaffolds: FimH), N-terminal display OmpX EstA Lpp-OmpA scaffolds (based on EstA, FhuA AIDA-I Invasin N AIDA-I, IgA protease), C-ter- N C FliTrx C IgA protease minal display scaffolds (based FimH on Lpp-OmpA, Invasin), and combination of N-terminal and C-terminal display using - - circularly permuted OmpX G bacteria G bacteria G+ bacteria: (CPX) could be chosen. For N-terminal C-terminal display via Gram-positive bacteria, N C display N C display covalently foreign proteins could be using using N anchored displayed via covalently CPX CPX proteins anchored proteins including C SPA SPA and FnBTB. FnBTB 2 Construction and screening of display library The protein or peptide library is constructed using recombi- nant DNA technology and displayed on bacteria. The bacterial library can be Library DNA labeled with fluorescently Lower-affinity variants Cloning Lower-affinity labeled target protein variants (antigen), or incubated with biotin-tagged target protein and streptavidin-magnetic beads. Target-binding bacte- ria are isolated using fluores- FACS MACS cence-activated cell sorting Highest-affinity Plasmid constructs Highest-affinity (FACS) or magnetic-activated variant variant cell sorting (MACS). The selection cycle is generally repeated, and selected bacte- ria are typically amplified by growth in between the cycles. E. coli Finally, isolated binders are screened using whole-cell Amplification Amplification ELISA-based methods, identi- fied using DNA sequencing and subcloned for soluble production and detailed characterization. -

Cell Surface Display
40 Cell Surface Display Sharadwata Pan and Michael K. Danquah Monash University Australia 1. Introduction The manipulation of the cell surfaces of prokaryotes (mainly bacteria) and eukaryotes (such as Yeast) has manifested to be an area of stupendous ongoing research, with intelligent widespread applications spanning different arenas of biological sciences (Charbit et al., 1988; Cruz et al., 2000; Francisco et al., 1993; Götz, 1990; Jostock & Dübel, 2005; Keskinkan et al., 2004; Kotrba et al., 1999; Lee & Schnaitman, 1980; Liljeqvist et al., 1997; Martineau et al., 1991; Mizuno et al., 1983; Sousa et al., 1996; Taschner et al., 2002; Wernérus & Ståhl, 2004; Willett et al., 1995; Xu & Lee, 1999). Till date, majority of the surface display systems developed for Gram-negative bacteria involve introducing external peptides into surface-approachable loops of naturally displayed proteins. This sometimes put extreme size restrictions on the displayed components (Wernérus & Ståhl, 2004). However, this problem is more or less resolved since larger proteins could be inserted through some recently developed bacterial display systems for Gram-negative bacteria (Charbit et al., 1988; Cruz et al., 2000; Lee & Schnaitman, 1980; Mizuno et al., 1983; Xu & Lee, 1999). Thanks to some tireless research, it is now evident that the structural properties of the cell wall in Gram-positive bacteria, i.e. the thick peptidoglycan layer, make them suitable candidates for strict laboratory procedures and demanding field applications (Jostock & Dübel, 2005). On the other hand, lower transformation efficiency has been a significant disadvantage of using Gram-positive bacteria (Wernérus & Ståhl, 2004), considering if someone is working with surface-displayed conjunctional libraries for affinity-based selections. -

UNIT – I - Fundamentals of Genomics and Proteomics– SBI1309
Genome organization and sequencing SCHOOL OF BIO AND CHEMICAL ENGINEERING DEPARTMENT OF BIOTECHNOLOGY UNIT – I - Fundamentals of Genomics and Proteomics– SBI1309 1 Genome organization and sequencing Organization of prokaryotic and eukaryotic genomes Prokaryotic Usually circular Smaller Found in the nucleoid region Less elaborately structured and folded Eukaryotic Complexed with a large amount of protein to form chromatin Highly extended and tangled during interphase Found in the nucleus The current model for progressive levels of DNA packing: Nucleosome basic unit of DNA packing formed from DNA wound around a protein core that consists of 2 copies each of the 4 types of histone (H2A, H2B, H3, H4)] A 5th histone (H1) attaches near the bead when the chromatin undergoes the next level of packing 30 nm chromatin fiber next level of packing; coil with 6 nucleosomes per turn the 30 nm chromatin forms looped domains, which are attached to a nonhistone protein scaffold (contains 20,000 – 100,000 base pairs) Looped domains attach to the inside of the nuclear envelope the 30 nm chromatin forms looped domains, which are attached to a nonhistone protein scaffold (contains 20,000 – 100,000 base pairs) 2 Genome organization and sequencing 3 Genome organization and sequencing Histones influence folding in eukaryotic DNA. Histones small proteins rich in basic amino acids that bind to DNA, forming chromatin Contain a high proportion of positively charged amino acids which bind tightly to the negatively charged DNA Heterochromatin Chromatin