1 United States District Court for the District Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Uniqure N.V. Paasheuvelweg 25A 1105BP Amsterdam the Netherlands +1-339-970-7000
uniQure N.V. Paasheuvelweg 25a 1105BP Amsterdam The Netherlands +1-339-970-7000 NOTICE OF EXTRAORDINARY GENERAL MEETING OF SHAREHOLDERS To be held on September 14, 2017 To the Shareholders of uniQure N.V.: Notice is hereby given that an Extraordinary General Meeting of Shareholders (the “Extraordinary Meeting”) of uniQure N.V., a public company with limited liability ( naamloze vennootschap ) under the laws of the Netherlands (the “Company,” “uniQure,” and “we”), will be held on September 14, 2017, at 9:30 a.m., Central European Summer Time, at the Company’s principal executive offices located at Paasheuvelweg 25a, 1105BP Amsterdam, the Netherlands, for the following purposes: I. Opening and announcements; II. Appointment of Jeremy P. Springhorn, Ph.D. as a non-executive director (voting proposal no. 1); III. Appointment of Madhavan Balachandran as a non-executive director (voting proposal no. 2); IV Any other business that may properly come before the meeting or any adjournment of the meeting; and V. Closing of the meeting. Each person authorized to attend the Extraordinary Meeting may inspect the Agenda at the office of uniQure. Our Board of Directors (our “Board”) recommends that you vote “FOR” each of the voting proposals noted above. The record date is set at the close of business on August 17, 2017 EST and, therefore, only the Company’s shareholders of record at the close of business on August 17, 2017 EST are entitled to receive this notice (this “Notice”) and to vote at the Extraordinary Meeting and any adjournment thereof. Only shareholders who have given notice in writing to the Company by September 12, 2017 of their intention to attend the Extraordinary Meeting in person are entitled to attend the Extraordinary Meeting in person. -

In the United States District Court for the District Of
IN THE UNITED STATES DISTRICT COURT FOR THE DISTRICT OF DELAWARE FOREST LABORATORIES, LLC and ) FOREST LABORATORIES ) HOLDINGS, LTD., ) ) Plaintiffs, ) ) v. ) Civ.No.14-1119-SLR ) SIGMAPHARM LABORATORIES, LLC, ) et al., ) ) Defendants. ) Jack B. Blumenfeld, Esquire and Maryellen Noreika, Esquire of Morris, Nichols, Arsht & Tunnell LLP, Wilmington, Delaware. Counsel for Plaintiffs. Of Counsel: Howard W. Levine, Esquire, Sanya Sukduang, Esquire, Jonathan R. Davies, Esquire, Courtney B. Gasp, Esquire, and Charles E. Lipsey, Esquire of Finnegan, Henderson, Farabow, Garrett & Dunner LLP. John C. Phillips, Esquire, David A. Bilson, Esquire and Megan C. Haney of Phillips, Goldman, Mclaughlin & Hall, P.A., Wilmington, Delaware. Counsel for Defendant Sigmapharm Laboratories, LLC. Of Counsel: Anthony G. Simon, Esquire, Anthony R. Friedman, Esquire, Benjamin R. Askew, Esquire, and Michael P. Kella, Esquire of The Simon Law Firm, P.C. Karen Elizabeth Keller, Esquire and Jeffrey Thomas Castellano, Esquire of Shaw Keller, LLP, Wilmington, Delaware. Counsel for Defendants Hikma Pharmaceuticals LLC, Hikma Pharmaceuticals PLC, and West-Ward Pharmaceutical Corp. Of Counsel: lmron T. Aly, Esquire, Joel M. Wallace, Esquire, and Helen H. Ji, Esquire of Schiff Hardin LLP. Richard D. Kirk, Esquire, Stephen B. Brauerman, Esquire and Sara E. Bussiere, Esquire of Bayard, P.A., Wilmington, Delaware. Counsel for Defendant Breckenridge Pharmaceutical, Inc. Of Counsel: Beth D. Jacob, Esquire, Clifford Katz, Esquire, and Malavika A. Rao, Esquire of Kelley, Drye & Warren LLP. Karen Pascale, Esquire and Pilar G. Kraman, Esquire of Young, Conaway, Stargatt & Taylor, LLP, Wilmington, Delaware. Counsel for Defendants Alembic Pharmaceuticals Ltd., Alembic Global Holding SA and Alembic Pharmaceuticals, Inc. Of Counsel: Steven J. Lee, Esquire, Michael K. -

Preclinical Development & IND Filing: Nuts, Bolts, Best Practices
“Preclinical Development & IND Filing: Nuts, Bolts, Best Practices and Regulatory Aspects.” Speakers: Amit Kalgutkar (Pfizer), Chandra Prakash (Agios), Sanjeev Thohan (Novartis), Li- Chun Wang (Takeda), Wei Yin (Biogen) Organizers: Sanjeev Thohan and Chandra Prakash Date: 6/29/2017 Time: 8:30 am – 5.00 pm Location: Boston/Cambridge Area: Marriott Kendall Square, 50 Broadway, Cambridge MA 02142 Fees: $175 - Regular (before June 1st, 2017), $225 - Regular (after June 1st, 2017); $125 - Unemployed & Academic; $2000 - Major Sponsorship; $475 - Vendor Show Registration: www.PBSS.org Workshop Description: An investigational new drug application (IND) is an important milestone that marks the entry of a molecule into clinical development. Knowing the objectives, expectations and processes of assembling an IND is a key to not only successful filing, but also a promising clinical development path forward. Often there are cases where too many of our “nice-to-have” studies crowd the package at the expense of critical study needs/issues. This can lead to significant delays in clinical developments with back-and-forth of Q&A sessions both internally and with regulatory agencies. As we have seen, the regulatory landscape is changing as rapidly as the industry innovates into new therapeutic modalities. Therefore, it is critical to keep up to date on regulatory requirements and the industry’s best practices in different aspects of the IND: non- clinical safety, PK, CMC, and clinical plans. In this workshop, our speakers who bring years of experience with multiple successful IND filings, will discuss systematically the preclinical studies required for small molecule IND’s as well as the nuts and bolts of putting together a high–quality IND package. -
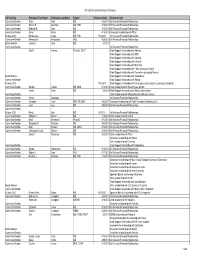
CB2013 Committee and Board Disclosureauditlist 9.10.13
2013-2014 Conflict of Interest Disclosure AST Activity Participant First Name Participant Last Name Degree DisclosureDate Disclsoure Item Committee Member Reza Abdi MD 4/22/2013 No Relevant Financial Relationships Committee Member Sameh R. Abul-Ezz MD, PhD 4/16/2013 No Relevant Financial Relationships Committee Member Deborah B. Adey MD 4/15/2013 No Relevant Financial Relationships Committee Member Enver Akalin MD 4/16/2013 Consultant relationship with Pfizer Fellows 2013 Maria-Luisa Alegre MD, PhD 7/12/2013 No Relevant Financial Relationships Committee Member Alessandrini Alessandro PhD 4/29/2013 No Relevant Financial Relationships Board Member James S. Allan MD 5/3/2013 Committee Member No Relevant Financial Relationships Rita R. Alloway PharmD, FCCP Grant Support relationship with Astellas Grant Support relationship with BMS Grant Support relationship with Novartis Grant Support relationship with Lifecycle Grant Support relationship with Millenium Grant Support relationship with Pfizer (previously Wyeth) Grant Support relationship with Genentech (previously Roche) Board Member Grant Support relationship with Viropharma Committee Member Grant Support relationship with Alexion Fellows 2013 7/18/2013 Grant Support relationship with Sanofi (previously Genzyme, previously Sangstat) Committee Member Sandra Amaral MD, MHS 6/18/2013 Other relationship with Bristol Myers-Squibb Hatem Amer MD 5/6/2013 Grant Support relationship with Abbott Laborartories. Committee Member Other relationship with Massacheussetts Medical Society. Committee Member William Applegate No Relevant Financial Relationships Committee Member Alexander Aussi BSN, RN, MBA 4/22/2013 Consultant relationship with Total Transplant Advantage, LLC Committee Member Jamil Azzi MD 4/30/2013 No Relevant Financial Relationships Committee Member Fellows 2013 Mark L. -

Assessment Report COVID-19 Vaccine Astrazeneca EMA/94907/2021
29 January 2021 EMA/94907/2021 Committee for Medicinal Products for Human Use (CHMP) Assessment report COVID-19 Vaccine AstraZeneca Common name: COVID-19 Vaccine (ChAdOx1-S [recombinant]) Procedure No. EMEA/H/C/005675/0000 Note Assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted. Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2021. Reproduction is authorised provided the source is acknowledged. Table of contents 1. Background information on the procedure .............................................. 7 1.1. Submission of the dossier ..................................................................................... 7 1.2. Steps taken for the assessment of the product ........................................................ 9 2. Scientific discussion .............................................................................. 12 2.1. Problem statement ............................................................................................. 12 2.1.1. Disease or condition ........................................................................................ 12 2.1.2. Epidemiology and risk factors ........................................................................... 12 2.1.3. Aetiology and pathogenesis ............................................................................. -

Surescripts, Llc As Amicus Curiae in Support of Petitioners in No
Nos. 19-508 and 19-825 In the Supreme Court of the United States ———————————— AMG CAPITAL MANAGEMENT, LLC, ET AL., Petitioners, v. FEDERAL TRADE COMMISSION, Respondent. ———————————— FEDERAL TRADE COMMISSION, Petitioner, v. CREDIT BUREAU CENTER, LLC, ET AL., Respondents. ———————————— ON WRITS OF CERTIORARI TO THE UNITED STATES COURTS OF APPEALS FOR THE SEVENTH AND NINTH CIRCUITS ———————————— BRIEF OF SURESCRIPTS, LLC AS AMICUS CURIAE IN SUPPORT OF PETITIONERS IN NO. 19-508 AND RESPONDENTS IN NO. 19-825 ———————————— ALFRED C. PFEIFFER, JR. ROMAN MARTINEZ LATHAM & WATKINS LLP Counsel of Record 505 Montgomery Street AMANDA P. REEVES Suite 2000 ALLYSON M. MALTAS San Francisco, CA 94111 DOUGLAS C. TIFFT BLAKE E. STAFFORD JAMES A. TOMBERLIN* LATHAM & WATKINS LLP 555 Eleventh Street, NW Suite 1000 Washington, DC 20004 (202) 637-2200 [email protected] Counsel for Amicus Curiae Surescripts, LLC TABLE OF CONTENTS Page TABLE OF AUTHORITIES ...................................... ii INTEREST OF AMICUS CURIAE ............................1 SUMMARY OF ARGUMENT .....................................3 ARGUMENT ...............................................................5 I. The FTC Has Increasingly Wielded Section 13(b) To Obtain Monetary Relief In Antitrust Cases ................................................5 II. The FTC’s Antitrust Authority Confirms That Section 13(b) Does Not Authorize Monetary Relief ..................................................22 CONCLUSION ..........................................................32 ii TABLE OF AUTHORITIES Page(s) CASES Apple Inc. v. Pepper, 139 S. Ct. 1514 (2019) .......................................... 13 Armstrong v. Exceptional Child Center, Inc., 575 U.S. 320 (2015) .............................................. 23 Bell Atlantic Corp. v. Twombly, 550 U.S. 544 (2007) .............................................. 26 In re Cardinal Health, Inc., No. 101-0006, 2015 WL 1849040 (F.T.C. Apr. 17, 2015) ........................ 19, 20, 28, 30 Credit Suisse Securities (USA) LLC v. Billing, 551 U.S. -
Astrazeneca-Oxford Vaccine Approved for Use in the U.K
P2JW366000-6-A00100-17FFFF5178F ****** THURSDAY,DECEMBER 31,2020~VOL. CCLXXVI NO.154 WSJ.com HHHH $4.00 DJIA 30409.56 À 73.89 0.2% NASDAQ 12870.00 À 0.2% STOXX 600 400.25 g 0.3% 10-YR. TREAS. À 3/32 , yield 0.926% OIL $48.40 À $0.40 GOLD $1,891.00 À $10.50 EURO $1.2300 YEN 103.21 Deadly Attack at Airport Targets New Yemen Government U.S. IPO What’s News Market Reaches Business&Finance Record nvestorspiled into IPOs Iat a record rate in 2020, with companies raising Total $167.2 billion via 454 of- ferings on U.S. exchanges this year through Dec. 24. Few see signs of letup Few expect the euphoria after companies raise to wear off soon. A1 more than $167 billion Detenteisending in the global fight over tech taxes, despite pandemic with Franceresuming collec- tion of itsdigital-services tax BY MAUREEN FARRELL and the U.S. poised to retali- atewith tariffs.Other coun- Defying expectations,inves- tries areset to join the fray. A1 S tors piled intoinitial public of- China finished 2020 PRES feringsatarecordrateiN with a 10th consecutive TED 2020, and few expect the eu- month of expansion in its CIA phoria to wear off soon. manufacturing sector. A7 SO Companies raised $167.2 AS TheEUand China agreed TENSIONS HIGH: People fled after an explosion Wednesday at the airport in Aden, Yemen, moments after members of the billion through 454 offerings in principle on an invest- country’s newly sworn-in cabinet arrived. At least 22 people were killed, but all the members of the cabinet were safe. -

United States District Court for the District of Columbia
Case 1:12-cv-00472-BAH Document 42 Filed 07/05/12 Page 1 of 49 UNITED STATES DISTRICT COURT FOR THE DISTRICT OF COLUMBIA ASTRAZENECA PHARMACEUTICALS LP, Plaintiff, v. Civil Action No. 12-00472 (BAH) Judge Beryl A. Howell FOOD AND DRUG ADMINISTRATION, et al., Defendants. AMENDED MEMORANDUM OPINION Plaintiff AstraZeneca Pharmaceuticals LP (“AstraZeneca”) has manufactured the drug quetiapine fumarate (“quetiapine”) under the brand name Seroquel® (“Seroquel”) since 1997 without generic competition. AstraZeneca brought this lawsuit, which presents a question of statutory interpretation, against the Food and Drug Administration, Margaret A. Hamburg, M.D., Commissioner of Food and Drugs, and Kathleen Sebelius, Secretary of Health and Human Services (collectively, “the FDA”), to challenge the FDA’s approval, on March 27, 2012, of generic versions of Seroquel. See Complaint, ECF No. 1 (“Compl.”), ¶ 3. AstraZeneca believes that, under the plain language of 21 U.S.C. § 355(j)(5)(F)(iv), codifying Section 505(j)(5)(F)(iv) of the Federal Food, Drug, and Cosmetic Act (“FDCA”), it is entitled to total market exclusivity until December 2, 2012 for the safety information encapsulated in “Table 2,” which was approved for all Seroquel labels on December 2, 2009 and must be included on the labels of all generic versions of quetiapine. Based upon this belief, AstraZeneca seeks a judgment that the FDA’s recent approval of generic versions of quetiapine, while AstraZeneca retains exclusivity over Table 2, violated AstraZeneca’s exclusivity rights and was arbitrary, capricious, and contrary to law. 1 Case 1:12-cv-00472-BAH Document 42 Filed 07/05/12 Page 2 of 49 Pending before the Court are Cross-Motions for Summary Judgment filed by AstraZeneca, ECF No. -

Federal Register/Vol. 84, No. 232/Tuesday, December 3, 2019
Federal Register / Vol. 84, No. 232 / Tuesday, December 3, 2019 / Notices 66191 the Assistant Attorney General, patterns, devices, manufacturing filed with and accepted, subject to final developed the HSR Rules and the processes, or customer names. approval, by the Commission, has been corresponding Notification and Report placed on the public record for a period Form. Heather Hippsley, of thirty (30) days. The following On September 11, 2019, the Deputy General Counsel. Analysis to Aid Public Comment Commission sought comment on the [FR Doc. 2019–26075 Filed 12–2–19; 8:45 am] describes the terms of the consent reporting requirements associated with BILLING CODE 6750–01–P agreement and the allegations in the the HSR Rules and corresponding complaint. An electronic copy of the Notification and Report Form. 84 FR full text of the consent agreement 47951. No relevant comments were FEDERAL TRADE COMMISSION package can be obtained from the FTC received. Pursuant to the OMB [File No. 191 0061] Home Page (for November 15, 2019), on regulations, 5 CFR part 1320, that the World Wide Web, at https:// implement the PRA, 44 U.S.C. 3501 et Bristol-Myers Squibb Company and www.ftc.gov/news-events/commission- seq., the FTC is providing this second Celgene Corporation; Analysis of actions. opportunity for public comment while Agreement Containing Consent Orders You can file a comment online or on seeking OMB approval to renew the pre- To Aid Public Comment paper. For the Commission to consider existing clearance for those information your comment, we must receive it on or collection requirements. -

Shire Acquisition of Viropharma - Strategic Move to Strengthen Shire’S Rare Disease Business - Augments Already Strong Growth Prospects
Shire acquisition of ViroPharma - Strategic move to strengthen Shire’s Rare Disease business - Augments already strong growth prospects Flemming Ornskov, MD Chief Executive Officer Graham Hetherington Chief Financial Officer Our purpose We enable people with life-altering conditions to lead better lives. CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS Statements included in this communication that are not historical facts are forward-looking statements. Forward-looking statements involve a number of risks and uncertainties and are subject to change at any time. In the event such risks or uncertainties materialize, results could be materially adversely affected. The risks and uncertainties include, but are not limited to, that: •Shire’s proposed acquisition of ViroPharma may not be consummated due to the occurrence of an event, change or other circumstances that gives rise to the termination of the merger agreement; •a governmental or regulatory approval required for the proposed acquisition of ViroPharma may not be obtained, or may be obtained subject to conditions that are not anticipated, or another condition to the closing of the proposed acquisition may not be satisfied; •ViroPharma may be unable to retain and hire key personnel and/or maintain its relationships with customers, suppliers and other business partners pending the consummation of the proposed acquisition by Shire, or ViroPharma’s business may be disrupted by the proposed acquisition, including increased costs and diversion of management time and resources; •difficulties in integrating ViroPharma into Shire may lead to the combined company not being able to realize the expected operating efficiencies, cost savings, revenue enhancements, synergies or other benefits at the time anticipated or at all; and risks and uncertainties detailed from time to time in Shire’s or ViroPharma’s filings with the U.S. -

Cephalon Provigil
r* jfS* 1 ll JS 44 {Rev. 07/16) SHEET The JS 44 civil cover sheet and the information contained herein neither replace nor supplement the filing and service of pleadings or other papers as required by law, except as provided by local rules of court. This form, approved by the Judicial Conference of the United States in September 1974, is required for the use of the Clerk of Court for the purpose ofinitiating the civil docket sheet. (SEE INSTRUCTIONS ON NEXT PAGE OF THIS FORM.) 1. (a) PLAINTIFFS DEFENDANTS State of New York, et al. Cephalon, Inc. et al. (b) County of Residence of First Listed Plaintiff County of Residence of First Listed Defendant (EXCEPT IN U.S. PLAINTIFF CASES) (IN U.S. PLAINTIFF CASES ONLY) NOTE: IN LAND CONDEMNATION CASES, USE THE LOCATION OF THE TRACT OF LAND INVOLVED. (c) Attorneys (Firm Name, Address, and Telephone Number) Attorneys (If Known) Office of the Attorney General, Commonwealth of Pennsylvania, 14® Floor Jay Lefkowitz, Kirkland & Ellis, 601 Lexington Avenue, New York, NY 10022 Strawberry Square, Harrisburg, PA 17120(717)787-4530 (212)446-4800 II. BASIS OF JURISDICTION (Placean "X" in One Box Only) III. CITIZENSHIP OF PRINCIPAL PARTIES (Placean "X" inOne Box for Plaintiff (For Diversify Cases Only) and One Box for Defendant) • 1 U.S. Government ^ 3 Federal Question PTF DEF PTF DEF Plaintiff (U.S. Government Not a Party) Citizen of This State Q 1 • 1 Incorporated or Principal Place Q 4 HI 4 of Business In This State l~l 2 U.S. Government • 4 Diversity Citizen of Another State • 2 Q 2 Incorporated and Principal Place G 5 Q 5 Defendant (Indicate Citizenship of Parties in Item III) of Business In Another State Citizen or Subject of a Q3 D 3 Foreign Nation D 6 06 Foreign Country IV. -

Constituents & Weights
2 FTSE Russell Publications 19 August 2021 FTSE 100 Indicative Index Weight Data as at Closing on 30 June 2021 Index weight Index weight Index weight Constituent Country Constituent Country Constituent Country (%) (%) (%) 3i Group 0.59 UNITED GlaxoSmithKline 3.7 UNITED RELX 1.88 UNITED KINGDOM KINGDOM KINGDOM Admiral Group 0.35 UNITED Glencore 1.97 UNITED Rentokil Initial 0.49 UNITED KINGDOM KINGDOM KINGDOM Anglo American 1.86 UNITED Halma 0.54 UNITED Rightmove 0.29 UNITED KINGDOM KINGDOM KINGDOM Antofagasta 0.26 UNITED Hargreaves Lansdown 0.32 UNITED Rio Tinto 3.41 UNITED KINGDOM KINGDOM KINGDOM Ashtead Group 1.26 UNITED Hikma Pharmaceuticals 0.22 UNITED Rolls-Royce Holdings 0.39 UNITED KINGDOM KINGDOM KINGDOM Associated British Foods 0.41 UNITED HSBC Hldgs 4.5 UNITED Royal Dutch Shell A 3.13 UNITED KINGDOM KINGDOM KINGDOM AstraZeneca 6.02 UNITED Imperial Brands 0.77 UNITED Royal Dutch Shell B 2.74 UNITED KINGDOM KINGDOM KINGDOM Auto Trader Group 0.32 UNITED Informa 0.4 UNITED Royal Mail 0.28 UNITED KINGDOM KINGDOM KINGDOM Avast 0.14 UNITED InterContinental Hotels Group 0.46 UNITED Sage Group 0.39 UNITED KINGDOM KINGDOM KINGDOM Aveva Group 0.23 UNITED Intermediate Capital Group 0.31 UNITED Sainsbury (J) 0.24 UNITED KINGDOM KINGDOM KINGDOM Aviva 0.84 UNITED International Consolidated Airlines 0.34 UNITED Schroders 0.21 UNITED KINGDOM Group KINGDOM KINGDOM B&M European Value Retail 0.27 UNITED Intertek Group 0.47 UNITED Scottish Mortgage Inv Tst 1 UNITED KINGDOM KINGDOM KINGDOM BAE Systems 0.89 UNITED ITV 0.25 UNITED Segro 0.69 UNITED KINGDOM