Brief Review of Advances in Developments of Living Polymers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalysis Robert J
REVIEW ARTICLE PUBLISHED ONLINE: 24 JULY 2012 | DOI: 10.1038/NCHEM.1405 The progression of chiral anions from concepts to applications in asymmetric catalysis Robert J. Phipps, Gregory L. Hamilton and F. Dean Toste* Despite the tremendous advances of the past four decades, chemists are far from being able to use chiral catalysts to con- trol the stereoselectivity of any desired reaction. New concepts for the construction and mode of operation of chiral catalysts have the potential to open up previously inaccessible reaction space. The recognition and categorization of distinct approaches seems to play a role in triggering rapid exploration of new territory. This Review both reflects on the origins as well as details a selection of the latest examples of an area that has advanced considerably within the past five years or so: the use of chiral anions in asymmetric catalysis. Defining reactions as involving chiral anions is a difficult task owing to uncertainties over the exact catalytic mechanisms. Nevertheless, we attempt to provide an overview of the breadth of reactions that could reasonably fall under this umbrella. 9 ithin the broader field of science, organic chemistry is Of significantly greater acidity (pKa around 2–4) are chiral phos- often referred to as a mature field of study. In spite of this, phoric acids, commonly derived from BINOL (1,1′-bi-2-naphthol). Wnew avenues of inquiry seem to arise with relative fre- These catalysts have risen to prominence over the past decade quency. Perhaps we take it for granted that expansions of the field because their high acidity permits activation of a broad range of are sparked when one or two chemical laboratories uncover a new substrates3,10. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

DNA Sequence Selectivity of Guanine-N7 Alkylation by Three Antitumor Chloroethylating Agents
ICANCER RESEARCH 46, 1943-1947, April 19861 DNA Sequence Selectivity of Guanine-N7 Alkylation by Three Antitumor Chloroethylating Agents John A. Hartley,' Neil W. Gibson, Kurt W. Kohn, and William B. Mattes@ L.aboratory ofMolecular Pharmacology. Developmental Therapeutics Program, Division ofCancer Treatment, National Cancer institute, NiH, Bethesda, Maryland 20205 ABSTRACF (4). Both compounds are like C1EtNUs in that they produce The DNA sequenceselectivitiesof guanine-N7alkylation producedby delayed interstrand cross-linking in Mer (guanine-O@-alkyl three chloroethylating antitumor agents, I-(2-chloroethyl)-3-(cis-2-hy transferase deficient) human tumor cell lines, but not in Mer droxy)cyclohexyl-l-nitrosourea(cis-2-OH CCNU), 2-chloroethyl(meth cells, DNA-protein cross-linking in both cell types without ylsulfonyl)methanesulfonate,and 8-carhamoyl-3-(2-chloroethyl)imidazo delay, and selective killing of the Mer cells (5, 6). 15,l-dl-l,2,3,5-tetrazin-4(3H)-one (mitozolomide), were examined using We have recently investigated whether either of these agents a modificationof the Maxam and Gilbert sequencing technique. In a had a simpler alkylation chemistry than the C1EtNUs. One of region ofpBR322 DNA, 2-chloroethyl(methylsulfonyl)methanesulfonate the major alkylation routes of the C1EtNUs is the production produced approximately the same degree of alkylation at all guanines. of hydroxyethyl adducts in DNA (7). Such adducts are thought cis-2-OH CCNU, however,preferentiallyalkylated the middleguanines to have little importance in the expression ofantitumor activity in runs of three or more guanines the intensity of the reaction increased but may contribute to both the mutagenic and carcinogenic with the number of adjacent guanines in the DNA sequence. -
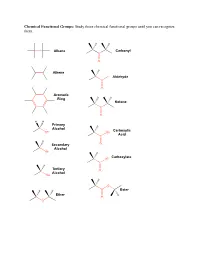
Study These Chemical Functional Groups Until You Can Recognize Them
Chemical Functional Groups: Study these chemical functional groups until you can recognize them. Alkane Carbonyl C O Alkene H Aldehyde C O Aromatic Ring Ketone C O H H Primary Alcohol Carboxylic OH OH Acid C H Secondary O Alcohol OH Carboxylate O C Tertiary O Alcohol OH O Ester Ether O O Primary Primary Ammonium Amine Ion NH3 NH2 Secondary Secondary Ammonium Amine Ion N N H H2 Tertiary Tertiary Ammonium Amine H N N Ion H Amide Imine N N O Thiol Thioester S SH O Sulfide S O Monophosphate P O O O O O Diphosphate P P O O O O O O O O Triphosphate P P P O O O O O O O 1. Draw the chemical structure of each amino acid for yourself. Identify every chemical functional group of each amino acid sidechain. ________________________________________________________________________________________________________ Hydrogen Bond Acceptors/Donors: “Polar molecules containing O-H and N-H bonds and the molecule HF have very large dipole moments and stronger than average dipole-dipole interactions. The hydrogen atoms in these molecules are bonded to small, highly electronegative atoms and have large partial positive charge. Therefore, the unequal distribution of the electrons in these molecules gives rise to large dipole moments. Because of its strength, the interaction between the partially positive hydrogen atom on one molecule and a partially negative O, N, or F atom with unshared pairs of electrons on an adjacent molecule merits special distinction: it is called a hydrogen bond” [Gilbert, Kirss, Foster, and Davies. Chemistry, 2nd Ed. 2009. pp. 473-474] 2. -

CYCLIC OXONIUM IONS and RELATED STRUCTURES* Whose
CYCLIC OXONIUM IONS AND RELATED STRUCTURES* BY ROBERT J. GARGIULO AND D. STANLEY TARBELL DEPARTMENT OF CHEMISTRY, VANDERBILT UNIVERSITY Communicated October 28, 1968 Abstract and Summary.-This paper describes the preparation by several methods of trialkyloxonium ions containing five-membered rings and the study of their structures by observing the nuclear magnetic resonance (NMR) spectra in several solvents. This provides independent evidence for these compounds, whose existence has been shown by kinetic, preparative, and stereochemical studies. The following SbCl6 oxonium salts have been prepared, isolated, and character- ized as crystalline solids: the 1,2,5-trimethyltetrahydrofuranium (Ha), the 1,2,2,5-tetramethyl (JIb), the trans-l-methylperhydrobenzofuranium (IVa), the trans-1,2-dimethyl (IVb), and the trans-1,2,2-trimethyl (IVc) compounds. The trifluoroacetate salts IIc, IId, IVd, and LWe have been made in acid solution, and all the salts II and IV have been characterized by their NMR spectra as cyclic- oxonium salts, with the charged 1-CH3 group downfield from an uncharged -OCH3. Spectra of all the SbCl6 salts in nitrobenzene solution are reported. Further evidence for the cyclic structure is given by the identification of 2,2,5- trimethyltetrahydrofuran (VIII) from the action of pyridine and lithium chloride on the 1,2,2,5-tetramethyltetrahydrofuranium salt JIb. Solvolysis of this salt by ethanol gave 2-ethoxy-2-methyl-5-methoxyhexane (VII) by attack at the tertiary carbon of the oxonium ion. The structure of VII was established by its synthesis and that of the other possible isomer. The trifluoroacetate oxonium ion (IVe) gave, after three days in 5 per cent sulfuric-trifluoroacetic acid, trans-2,2- dimethylperhydrobenzofuran and methyl trifluoroacetate. -

Tricyclic Oxonium Tamed
RESEARCH HIGHLIGHTS Nature Reviews Chemistry | https://doi.org/10.1038/s41570-019-0150-y | Published online 13 November 2019 NATURAL PRODUCT SYNTHESIS could only be characterized at low temperature. However, 1H and 13C NMR spectra are indicative of its Tricyclic oxonium tamed formation with large downfield shifts of the signals for the three Natural products or secondary formal positive charge on oxygen carbons adjacent to the oxygen and metabolites have hugely diverse (a trialkyloxonium ion) proposed for the protons attached to them. We decided structures. Simple-looking linear for other biosyntheses have proved In addition, a new cross-peak was to try to precursors can, via highly reactive far more elusive. observed in the 1H–13C correlation intermediates, fold up into a variety “The structures of a number spectrum, indicating the formation investigate of complex 3D structures. Writing of Laurencia natural products had of a new transannular bond. 1H and whether these in the Journal of the American originally been misassigned,” explains 13C chemical shifts calculated somewhat Chemical Society, Jonathan Burton Burton. “Our work with Rob Paton on using density functional theory also exotic looking and co-workers from the University calculating the NMR spectra for the favoured the assigned structures and of Oxford, in collaboration with proposed structures of these natural formation of the oxonium ions. species might Robert Paton at Colorado State products led us to earlier proposals of Four different oxonium ions really exist University, describe the synthesis and tricyclic oxonium species. We decided derived from the diastereomers characterization of tricyclic oxonium to try to investigate whether these of laurediol were prepared and intermediates and their conversion somewhat exotic looking species characterized — the formation of into ten different natural products might really exist.” one of these, from 3Z, S,S-laurediol originally isolated from algae of the In the proposed biosyntheses, is pictured. -

Silane/Iodine-Based Cleavage of Esters and Ethers Under Neutral
Proc. Nati. Acad. Scd. USA Vol. 75, No. 1, pp. 4-6, January 1978 Chemistry Silane/iodine-based cleavage of esters and ethers under neutral conditions* (ester cleavage/ether cleavage/dealkylation/iodotrimethylsilane/phenyltrimethylsilane-iodine) TSE-LOK HO AND GEORGE A. OLAH Institute of Hydrocarbon Chemistry, Department of Chemistry, University of Southern California, Los Angeles, California 90007 Contributed by George A. Olah, September 26, 1977 ABSTRACT New, efficient cleavage of carboxylic esters physical constants and spectral data that were in good agree- with iodotrimethylsilane or a mixture of phenyl imethylsilane ment with literature data or those of authentic samples. and iodine is described. Essentially neutral conditions can be maintained throughout the reactions. Ethers can be dealkylated Cleavage of Esters and Ethers by Phenyltrimethylsil- by the latter method in high yields. The mechanism of the ane/lodine. A mixture of the ester or ether (5 mmol), phen- cleavage reactions is considered to include six-membered ring yltrimethylsilane (1.0 g, 7.5 mmol), and iodine (1.91 g, 15 mg homopolar transition states. atom) was heated to 1100 for 2-6 hr (see Tables 3 and 4). It was then cooled and quenched with water, and the organic material The most commonly used and convenient method for pro- was extracted with benzene and washed with dilute sodium tecting carboxylic acids is esterification. In view of the frequent hydroxide. The aqueous phase was acidified, deiodinated with incompatibility, during ester hydrolysis, of alkaline conditions Na2S203, and extracted with benzene. Pure carboxylic acids with many sensitive functional groups, esters that permit and phenols were isolated from the benzene solution. -

Aldehydes Can React with Alcohols to Form Hemiacetals
340 14 . Nucleophilic substitution at C=O with loss of carbonyl oxygen You have, in fact, already met some reactions in which the carbonyl oxygen atom can be lost, but you probably didn’t notice at the time. The equilibrium between an aldehyde or ketone and its hydrate (p. 000) is one such reaction. O HO OH H2O + R1 R2 R1 R2 When the hydrate reverts to starting materials, either of its two oxygen atoms must leave: one OPh came from the water and one from the carbonyl group, so 50% of the time the oxygen atom that belonged to the carbonyl group will be lost. Usually, this is of no consequence, but it can be useful. O For example, in 1968 some chemists studying the reactions that take place inside mass spectrometers needed to label the carbonyl oxygen atom of this ketone with the isotope 18 O. 16 18 By stirring the ‘normal’ O compound with a large excess of isotopically labelled water, H 2 O, for a few hours in the presence of a drop of acid they were able to make the required labelled com- í In Chapter 13 we saw this way of pound. Without the acid catalyst, the exchange is very slow. Acid catalysis speeds the reaction up by making a reaction go faster by raising making the carbonyl group more electrophilic so that equilibrium is reached more quickly. The the energy of the starting material. We 18 also saw that the position of an equilibrium is controlled by mass action— O is in large excess. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

An Automated Ion Library Generator for Data Independent Acquisition Mass Spectrometry Analysis of Peptides and Glycopept
bioRxiv preprint doi: https://doi.org/10.1101/696518; this version posted July 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. DIALib: an automated ion library generator for data independent acquisition mass spectrometry analysis of peptides and glycopeptides Toan K. Phung1*, Lucia F Zacchi2*, and Benjamin L. Schulz1,2¥ 1 School of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, Queensland, 4072, Australia. 2 ARC Training Centre for Biopharmaceutical Innovation, The University of Queensland, St. Lucia, QLD 4072, Australia. Running title: Automated construction of ion libraries for DIA ¥To whom correspondence should be addressed: Benjamin L. Schulz, School of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, Queensland, 4072, Australia. Tel.: +0061-7-336-54875; E-mail: [email protected] *These authors contributed equally to this work Keywords: SWATH-MS, peptide library, glycoproteomics, N-linked glycosylation, O-linked glycosylation bioRxiv preprint doi: https://doi.org/10.1101/696518; this version posted July 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Data Independent Acquisition (DIA) Mass Spectrometry (MS) workflows allow unbiased measurement of all detectable peptides from complex proteomes, but require ion libraries for interrogation of peptides of interest. -

Electrophilic Addition Reactions.Pdf
Electrophilic Addition Reactions Electrophilic addition reactions are an important class of reactions that allow the interconversion of C=C and C≡C into a range of important functional groups. Conceptually, addition is the reverse of elimination What does the term "electrophilic addition" imply ? A electrophile, E+, is an electron poor species that will react with an electron rich species (the C=C) An addition implies that two systems combine to a single entity. Depending on the relative timing of these events, slightly different mechanisms are possible: • Reaction of the C=C with E+ to give a carbocation (or another cationic intermediate) that then reacts with a Nu- • Simultaneous formation of the two σ bonds The following pointers may aid your understanding of these reactions: • Recognise the electrophile present in the reagent combination • The electrophile adds first to the alkene, dictating the regioselectivity. • If the reaction proceeds via a planar carbocation, the reaction is not stereoselective • If the two new σ bonds form at the same time from the same species, then syn addition is observed • If the two new σ bonds form at different times from different species, then anti addition is observed Hydrogenation of Alkenes Reaction Type: Electrophilic Addition Summary • Alkenes can be reduced to alkanes with H2 in the presence of metal catalysts such as Pt, Pd, Ni or Rh. • The two new C-H σ bonds are formed simultaneously from H atoms absorbed into the metal surface. • The reaction is stereospecific giving only the syn addition product. • This reaction forms the basis of experimental "heats of hydrogenation" which can be used to establish the stability of isomeric alkenes.