Ion Pair Conductivity Model and Its Application for Predicting Conductivity in Non-Polar Systems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Chemistry of the Carbon-In--Pulp Process
THE CHEMISTRY OF THE CARBON-IN--PULP PROCESS Michael David Adams A Thesis submitted to the Faculty of Science University of the Witwatersrand, Johannesburg in fulfilment of the requirements for the degree of Doctor of Philosophy Johannesburg 1989 ABSTRACT Several conflicting theories of the adsorption of aurocyanide onto activated carbon presently exist. To resolve the mechanism, adsorption and elution of aurocyanide are examined by several techniques, including Mossoauer spectroscopy, X-ray photoelectron spectro scopy, X-ray diffractometry, Fourier Transform Infrared spectrophotometry, ultraviolet-visible spectrophotometry and scanning electron microscopy. The evidence gathered indicates that, under normal plant conditions, aurocyanide is extracted onto activated carbon in the form of an ion pair M n* [Au(CN) 2 3 n , and eluted by hydroxide or cyanide. The hydroxide or cyanide ions react with the carbon surface, rendering it relatively hydrophilic with a decreased affinity for neutral species. Additional adsorption mechanisms are shown to operate under other conditions of ionic strength, pH, and temperature. The poor agreement in the literature regarding the mechanism of adsorption of aurocyanide onto activated carbon is shown to be due to the fact that different mechanisms operate under different experimental conditions. The AuCN produced on the carbon surface by acid treatment is shown to react with hydroxide ion via the reduction of AuCN to metallic gold with formation of Au (CN) 2 , and the oxidation of cyanide to cyanate. Other species, such as An(CN)5 and Ag(CN)g adsorb onto activated carbon by a similar mechanism to that postulated for Au( C N ) 2 . Ion association of MAu(CN ) 2 salts in aqueous solution is demonstrated by * aans of potentiometric titration and conductivity measurements, and various associated species of KAu(CN), salts are shown to occur in organic solvents by means of infrared spectrophoteaietric and distribution measurements. -
CRC Handbook of Chemistry and Physics, 91Th Edition
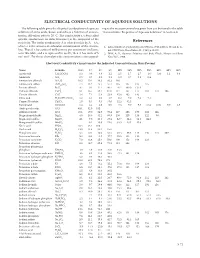
ELECTRICAL CONDUCTIVITY OF AQUEOUS SOLUTIONS The following table gives the electrical conductivity of aqueous ing to the mass percent values given here can be found in the table solutions of some acids, bases, and salts as a function of concen- “Concentrative Properties of Aqueous Solutions” in Section 8 . tration . All values refer to 20 °C . The conductivity κ (often called specific conductance in older literature) is the reciprocal of the resistivity . The molar conductivity Λ is related to this by Λ = κ/c, References where c is the amount-of-substance concentration of the electro- 1 . CRC Handbook of Chemistry, and Physics, 70th Edition, Weast, R . C ., lyte . Thus if κ has units of millisiemens per centimeter (mS/cm), Ed ., CRC Press, Boca Raton, FL, 1989, p . D-221 . as in this table, and c is expressed in mol/L, then Λ has units of S 2 . Wolf, A . V ., Aqueous Solutions and Body Fluids, Harper and Row, cm2 mol-1 . For these electrolytes the concentration c correspond- New York, 1966 . Electrical Conductivity κ in mS/cm for the Indicated Concentration in Mass Percent Name Formula 0.5% 1% 2% 5% 10% 15% 20% 25% 30% 40% 50% Acetic acid CH3COOH 0 .3 0 .6 0 .8 1 .2 1 .5 1 .7 1 .7 1 .6 1 .4 1 .1 0 .8 Ammonia NH3 0 .5 0 .7 1 .0 1 .1 1 .0 0 .7 0 .5 0 .4 Ammonium chloride NH4Cl 10 .5 20 .4 40 .3 95 .3 180 Ammonium sulfate (NH4)2SO4 7 .4 14 .2 25 .7 57 .4 105 147 185 215 Barium chloride BaCl2 4 .7 9 .1 17 .4 40 .4 76 .7 109 .0 137 .0 Calcium chloride CaCl2 8 .1 15 .7 29 .4 67 .0 117 157 177 183 172 106 Cesium chloride CsCl 3 .8 7 .4 13 .8 -

Topic2270 Ion Association the Term ' Strong Electrolyte' Has a Long And
Topic2270 Ion Association The term ‘ strong electrolyte’ has a long and honourable history in the development of an understanding of the properties of salt solutions. This term describes salt solutions where each ion contributes to the properties almost independently of all other ions in a given solution. The word ‘almost’ signals that the properties of a given salt solution are determined in part by charge –charge interactions between ions through the solvent separating ions in solution. Otherwise the ions can be regarded as free. Such is the case for aqueous salt solutions at ambient temperatures and pressures prepared + - + - using 1:1 salts such as Na Cl , Et4N Br … However with decrease in relative permittivity of the solvent, the properties of salt solutions indicate that not all the ions can be regarded as free; a fraction of the ions are associated. For dilute salts solutions in apolar solvents such as propanone a fraction of the salt is described as being present as ion pairs formed by association of cations and anions. With further decrease in the permittivity of the solvent higher clusters are envisaged; e.g. triple ions, quadruple ions…. Here we concentrate attention on ion pair formation building on the model proposed by N. Bjerrum [1,2]. The analysis identifies a given j ion in a salt solution as the reference ion such that at distant r from this ion the electric potential equals ψj whereby the potential energy ⋅ ⋅ ψ of ion i with charge number zi equals z i e j . The solvent is a structureless continuum and each ion is a hard non-polarisable sphere characterised by its charge, ⋅ z j e , and radius rj. -

Ion-Association a Review' Reagents
ANALYTICAL SCIENCES DECEMBER 1987, VOL. 3 479 Reviews Ion-Association Reagents A Review' Kyoji TBEI Faculty of Liberal Arts and Science, Okayama University of Science, Ridaicho, Okayama 700 Ion-association reagents are widely used for analytical chemistry. The ion-associable cation or anion should be univalent, bulky and charge-dispersed. The reagents are classified by their shape: plane, sphere, chain and polymer types. The ion-associable cation and anion can combine with each other to form the ion-associate in water. This phenomenon has been used for gravimetric and volumetric analyses and for extraction-spectrophoto- metry of a trace component. Several examples of specific and sensitive extraction-spectrophotometries are illustrated. The polymer type reagent can react with the counter-charged poly-electrolyte to form a precipitate quantitatively, and it can be used for colloid titration. Metachromasy phenomenon is based on the formation of the ion-associate of colored ion-associable cation or anion with counter-charged colorless ion-association reagents to show distinguished color changes. Keywords Ion-association reagents, charged quinone theory, extraction-spectrophotometry, colloid titration, metachromasy with ion-associable cations and the cationic one reacts 1 Introduction with ion-associable anions to form the respective ion- associates in an aqueous solution. When the Many kinds of organic reagents have been used for concentration of the ion-associate is relatively high in analytical chemistry. Most of the reagents are water, it is precipitated. This fact is used for chelating reagents. On the other hand, there is an ion- gravimetric or volumetric analysis. When it is diluted, association reagent which does not belong to the it is extracted into an organic solvent which is chelating reagent. -

Electrostatic and Relativistic Contributions to Ion-Pairing in Polyoxometalate Model Systems
“This document is the Accepted Manuscript version of a Published Work that appeared in final form in Phys. Chem. Chem. Phys. 2017, 19, 8715, copyright © The Royal Chemistry Society after peer review and technical editing by the publisher. To access the final edited and published work see DOI 10.1039/C6CP08454K. Electrostatic and relativistic contributions to ion-pairing in polyoxometalate model systems Dylan J. Sures, Stefano A. Serapian, Karoly Kozma, Pedro I. Molina, Carles Bo, and May Nyman Abstract Ion pairs and solubility related to ion-pairing in water influence many processes in nature and in synthesis including efficient drug delivery, contaminant transport in the environment, and self-assembly of materials in water. Ion pairs are difficult to observe spectroscopically because they generally do not persist unless extreme solution conditions are applied. Here we demonstrate two advanced techniques coupled with computational studies that quantify the persistence of ion-pairs in simple solutions and offer explanations for observed solubility trends. The system of study, (TMA,Cs)8[M6O19] (M=Nb,Ta), is a set of unique polyoxometalate salts whose water solubility increases with increasing ion-pairing, contrary to + 8- most ionic salts. The techniques employed to characterize Cs association with [M6O19] and related clusters in simple aqueous media are 133Cs NMR (nuclear magnetic resonance) quadrupolar relaxation rate and PDF (pair distribution function) from X-ray scattering. The NMR measurements consistently showed more extensive ion-pairing of Cs+ with the Ta-analogue than the Nb-analogue, although the electrostatics of the ions should be identical. Computational studies also ascertained more persistent Cs+- + [Ta6O19] ion pairs than Cs -[Nb6O19] ion pairs, and bond energy decomposition analyses determined relativistic effects to be the differentiating factor. -

Ion Association and Solvation in Dichloromethane of Tetrachloro- and Tetrabromoferrates(III) Compared with Simple Halides
Ion Association and Solvation in Dichloromethane of Tetrachloro- and Tetrabromoferrates(III) Compared with Simple Halides S. Bait, G. du Chattel, W. de Kieviet, and A. Tieleman Department of Inorganic Chemistry Free University, De Lairessestraat 174, Amsterdam, The Netherlands Z. Naturforsch. 33b, 745-749 (1978); received April 26, 1978 Ion Association, Dichloromethane, Tetrachloroferrate(III), Tetrabromoferrate(III), Hydrogen-bridge Formation Conductivity measurements are reported in dichloromethane for tetraethylammonium and tetraphenylarsonium salts of tetrachloro- and tetrabromoferrate(III) and halide ions (chiefly chloride and bromide). Analysis of the data in terms of the recent Fuoss equation has given the ion-pair association constants. The experimental values are close to the ones calculated from the electrostatic Fuoss-Eigen equation. The solvent acts as an acceptor by way of forming hydrogen-bonds to the simple halide ions and to a much less extent to the ferrates. Solvation of chloro- and bromoferrates(III) seems to be comparable, as deduced from the association constants and NMR paramagnetic line broadening. The phenyl groups of the tetraphenylarsonium ion seem to give ^-interaction with the solvent. Introduction proved to be a very good starting point, because it has negligible donor-properties [1, 2], but is a In the study of kinetics and mechanism of co- remarkably good solvent for ion-paired complexes. ordination compounds the emphasis seems to be In addition the symmetrical nature of the anion shifting from aqueous to non-aqueous solvents. In makes it in combination with symmetrical cations this connection a (semi)quantitative knowledge of a model case for treating ion-association with cur- donor and acceptor properties [1] of the various rent conductivity theories. -

Ii ION-PAIR BEHAVIOR BETWEEN POLYOXOMETALATES ANION
ION-PAIR BEHAVIOR BETWEEN POLYOXOMETALATES ANION AND ALKALI METAL CATION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirement for the Degree Master of Science Songtao Ye May 2018 ii ION-PAIR BEHAVIOR BETWEEN POLYOXOMETALATES ANION AND ALKALI METAL CATION Songtao Ye Thesis Approved Accepted: ______________________________ ____________________________ Advisor Dean of the College Dr. Tianbo Liu Dr. Eric Amis ______________________________ ____________________________ Committee Member Dean of the Graduate School Dr. Toshikazu Miyoshi Dr. Chand Midha ______________________________ ____________________________ Department Chair Date Dr. Colleen Pugh iii ABSTRACT Ion-pair behavior describes the partial association of oppositely charged ions in electrolyte solutions. Previous study mainly focused on the ion-pair behavior between simple ions, such as ion pairing in NaCl solution as well as ion-pair interactions in supramolecular complexes and biological associations. However, very few attentions have been placed on the solution system with particle sizes in between. Recently, a group of well-defined, huge anionic cluster named polyoxometalates (POMs) have been synthesized and well characterized. The size of POMs is around nanometer scale, which is exactly between simple ions and large colloids. The solution behavior for POMs is much different form simple electrolyte solutions or large colloids. As a result, it is interesting to study the ion-pair behavior for POMs in solution. Herein, ion-pairs between Lacunary Keggin type POMs and alkali metal cations are investigated. The result showed that ion-pairs are formed between alkali cations and the “pocket” area on the surface of Lacunary Keggin type POMs – K7PW11O39. Electrostatic interaction and the entropy gain during the solvation shell lost were considered major driving forces during the ion-pair formation. -

Electrochemical and Computational Study of Ion Association in the 3− Electroreduction of PW12O40 J
Article Cite This: J. Phys. Chem. C XXXX, XXX, XXX-XXX pubs.acs.org/JPCC Electrochemical and Computational Study of Ion Association in the 3− Electroreduction of PW12O40 J. M. Gomez-Gil,́ † E. Laborda,† J. Gonzalez,† A. Molina,*,† and R. G. Compton‡ † Departamento de Química Física, Facultad de Química, Regional Campus of International Excellence “Campus Mare Nostrum”, Universidad de Murcia, 30100 Murcia, Spain ‡ Department of Chemistry, Physical & Theoretical Chemistry Laboratory, Oxford University, South Parks Road, Oxford OX1 3QZ, United Kingdom *S Supporting Information ABSTRACT: Insights into ion pairing effects on the redox 3− properties of the Keggin-type polyoxotungstate PW12O40 are gained by combining electrochemical experiments and density functional theory (DFT) calculations. Such effects have been reported to affect the performance of these species as molecular electrocatalysts. Experimental square wave voltam- 3− metry (SWV) of the two-electron reduction of PW12O40 in 4− acetonitrile evidences that the reduced forms PW12O40 and 5− fi PW12O40 can be signi cantly stabilized by ion association. The strength and stoichiometry of the corresponding aggregates are estimated as a function of the nature of the cation (lithium, sodium, and tetramethylammonium) and the oxidation state of the polyoxometalate. The results obtained in combination with DFT enable us to examine the roles of the cation solvation and the charge number and distribution of the polyanions. 1. INTRODUCTION electrochemical and quantum-chemical approach. As will be discussed, the “apparent” relative stability in solution of PW3− Polyoxometalates (POMs) are anionic metal-oxide clusters that 4− 5− show particular electronic properties of great interest in a large and the reduced forms PW and PW can be elucidated from − number of areas,1 5 specifically in electrocatalysis (water voltammetric measurements in an aprotic medium (acetoni- oxidation,6,7 epoxidation of alkenes,8 bromate reduction,9 trile). -

Stepwise Association of Hydrogen Cyanide with the Pyridine and Pyrimidine Radical Cations and Protonated Pyridine Ahmed M
Virginia Commonwealth University VCU Scholars Compass Chemistry Publications Dept. of Chemistry 2014 Unconventional hydrogen bonding to organic ions in the gas phase: Stepwise association of hydrogen cyanide with the pyridine and pyrimidine radical cations and protonated pyridine Ahmed M. Hamid Virginia Commonwealth University M. Samy El-Shall Virginia Commonwealth University, [email protected] Rifaat Hilal King Abdulaziz University Shaaban Elroby King Abdulaziz University FSaoallodulwl ahthi Gs a. ndAz iazdditional works at: http://scholarscompass.vcu.edu/chem_pubs King Abdulaziz University Part of the Chemistry Commons Hamid, A. M., El-Shall, M. S., & Hilal, R., et al. Unconventional hydrogen bonding to organic ions in the gas phase: Stepwise association of hydrogen cyanide with the pyridine and pyrimidine radical cations and protonated pyridine. The Journal of Chemical Physics, 141, 054305 (2014). Copyright © 2014 AIP Publishing LLC. Downloaded from http://scholarscompass.vcu.edu/chem_pubs/66 This Article is brought to you for free and open access by the Dept. of Chemistry at VCU Scholars Compass. It has been accepted for inclusion in Chemistry Publications by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. THE JOURNAL OF CHEMICAL PHYSICS 141, 054305 (2014) Unconventional hydrogen bonding to organic ions in the gas phase: Stepwise association of hydrogen cyanide with the pyridine and pyrimidine radical cations and protonated pyridine Ahmed M. Hamid,1 M. Samy El-Shall,1,a) -

Electrochemistry Galvenic Cell Or Voltoic Cell 1. Identify the Location of Oxidation in an Electrochemical Cell. A) the Anode B)
Electrochemistry Galvenic cell or voltoic cell 1. Identify the location of oxidation in an electrochemical cell. a) the anode b) the cathode c) the electrode d) the salt bridge 2. Which one of the following is not a function of a salt bridge? a) To allow the flow of cations from one solution to the other b) To allow the flow of anions from one solution to the other c) To allow the electrons to flow from one solution to the other d) To maintain electrical neutrality of the two solutions. 3. If the salt bridge is removed suddenly from a working cell, the voltage a) Increases b) decreases c) Drops to zero d) May increase or decrease depending upon cell reaction 4. A cell is formed by the combination of a standard Mg electrode and a standard Zn electrode. Their reduction potentials are respectively –2.36V and ‐ 0.76V. The emf of the cell is a) +1.6V b) –1.6V b) +3.12V d) none of these 5. Copper has less SRP than silver (placed above silver in the electrochemical series). So if a copper electrode is combined with a silver electrode to form a cell, then the cell operates. a) Copper is oxidised b) silver is oxidized c) Silver gains electrons d) silver loses electrons. 6. The reduction potentials of three elements A, B and C are –2.37V, ‐0.76V, and +0V respectively. Then from the aqueous solution. a) C displaces hydrogen b) C is bases than A & B c) A displaces B but not C d) A displaces B and C. -

Thermodynamic Studies of Some Symmetrical Electrolyte's Solution
ISSN 1022-8594 2018 Jahangirnagar University Journal of Science JUJS Vol. 41, No.1, pp.87-98 Thermodynamic Studies of Some Symmetrical Electrolyte’s Solution in Aqueous-Organic Solvent Mixtures Md. Minarul Islam* and Md. Abdullah-Al-Mahmud Department of Chemistry, Jahangirnagar University, Savar, Dhaka-1342 Abstract Thermodynamic properties of the solutions of manganese sulphate (MnSO4), nickel sulphate (NiSO4), and zinc sulphate (ZnSO4) in aqueous-organic mixtures containing 5%, 10%, 15% and 20% (w/w) of ethanol have been measured at 250C in the concentration range of 2.0x10-4 to 12.0x10-4 mol dm-3. The standard Gibbs free energy, entropy and enthalpy changes were calculated from the conductance data using the values of ion-association constant by thermodynamic equations. The Gibbs free energy, entropy and enthalpy changes were investigated depending on the amount of ethanol percentage. 0 Gibbs energy of transfer (G t) from aqueous media to aqueous-organic mixtures of individual ions was also calculated in different solvent mixtures. The contributions of electrostatic and non-electrostatic part to the transfer free energy were also calculated from free energy of transfer. The values of Gibbs free energies were found negative for each electrolyte which is the sign of spontaneous association process. The values of entropies and enthalpies were found positive for each electrolyte over the entire range of solvent compositions. Keywords: Electrolytes, ion association constant, Gibbs free energy, entropy, enthalpy, Gibbs energy of transfer. Introduction Thermodynamic properties for the solutions of 2:2 electrolytes in aqueous and aqueous-organic solvent mixtures are of great interest from both theoretical and practical point of view. -

Superalkali–Alkalide Interactions and Ion Pairing in Low-Polarity Solvents
pubs.acs.org/JACS Article Superalkali−Alkalide Interactions and Ion Pairing in Low-Polarity Solvents René Riedel,* Andrew G. Seel,* Daniel Malko, Daniel P. Miller, Brendan T. Sperling, Heungjae Choi, Thomas F. Headen, Eva Zurek, Adrian Porch, Anthony Kucernak, Nicholas C. Pyper, Peter P. Edwards,* and Anthony G. M. Barrett* Cite This: J. Am. Chem. Soc. 2021, 143, 3934−3943 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: The nature of anionic alkali metals in solution is traditionally thought to be “gaslike” and unperturbed. In contrast to this noninteracting picture, we present experimental and computational data herein that support ion pairing in alkalide solutions. Concentration dependent ionic conductivity, dielectric spectroscopy, and neutron scattering results are consistent with the presence of superalkali−alkalide ion pairs in solution, whose stability and properties have been further investigated by DFT calculations. Our temperature dependent alkali metal NMR measurements reveal that the dynamics of the alkalide species is both reversible and thermally activated suggesting a complicated exchange process for the ion paired species. The results of this study go beyond a picture of alkalides being a “gaslike” anion in solution and highlight the significance of the interaction of the alkalide with its complex countercation (superalkali). ■ INTRODUCTION width. Considering that the alkali metals all possess quadrupolar nuclei, these features have been ascribed to the Anionic forms of the electropositive Group I metals, with the “ ” exception of lithium, can be generated in condensed phases.1 high shielding and high symmetry of an unperturbed gaslike anion in solution, with little to no interaction with its local Termed alkalides, these monoanions are chemically highly − environment.9 14 However, the high polarizability of the reducing and possess a diffuse, closed-shell ns2 configuration, alkalides and ready electron dissociation into solvated-electron resulting in an exceptionally high electronic polarizability.