Analysis of Hepatitis B Virus Genotype D in Greenland Suggests Presence of a Novel Subgenotype
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Greenland Explorer
GREENLAND EXPLORER Valleys and Fjords EXPEDITION IN BRIEF The Trip Overview Meet locals along the west coast of Greenland and experience traditional Inuit settlements Visit the Ilulissat Icefjord, a UNESCO World Heritage Site The west coast of Greenland is Europe’s final frontier, and sailing along it is Explore historic places from Norse the best way to sample its captivating history, enthralling wildlife and distinct and Viking eras culture. Explore places from the Norse and Viking eras, experience the Spot arctic wildlife, such as whales, birds and seals Ilulissat Icefjord—a UNESCO World Heritage Site— and visit two Greenland Cruise in a Zodiac to get up close to communities, encountering an ancient culture surviving in a modern world. glaciers, fjords, icebergs and more For trip inquiries, speak to our Polar Travel Advisers at 1. 844.205.0837 | Visit QuarkExpeditions.com for more details or get a free quote here. and geography of Greenland, your next Itinerary stop. Join expedition staff on deck and on the bridge as they look out for whales and seabirds, get to know your fellow Ban Bay GREENLAND DAY 1 | ARRIVE IN guests or simply take in the natural REYKJAVIK, ICELAND ARCTIC beauty that surrounds you. CIRCLE Arrive in the Icelandic capital in the Eqip Sermia Ilulissat morning and make your way to your DAY 4 | EAST GREENLAND Sisimiut Kangerlussuaq Experience a true arctic ghost town Itilleq included hotel. You will have the day Scoresby Sund to explore the city on your own. In when we visit the abandoned settlement Nuuk of Skjoldungen, where inhabitants the evening, join us at your hotel for a Skjoldungen Denmark Strait welcome briefing. -

(Laksebugt), South-West Disko, Greenland – Implications for Sea-Level Reconstructions
Beach-ridge architecture constrained by beach topography and ground-penetrating radar, Itilleq (Laksebugt), south-west Disko, Greenland – implications for sea-level reconstructions PRISCILA E. SOUZA, AART KROON & LARS NIELSEN Souza, P.E., Kroon, A. & Nielsen, L. 2018. Beach-ridge architecture constrained by beach topography and ground-penetrating radar, Itilleq (Laksebugt), south-west Disko, Greenland – implications for sea-level reconstructions. © 2018 by Bulletin of the Geological Society of Denmark, Vol. 66, pp. 167–179. ISSN 2245-7070. (www.2dgf.dk/publikationer/bulletin). https://doi.org/10.37570/bgsd-2018-66-08 Detailed topographic data and high-resolution ground-penetrating radar (GPR) reflection data are presented from the present-day beach and across successive raised beach ridges at Itilleq, south-west Disko, West Greenland. In the western part of the study area, the present low-tide level is well Received 24 November 2017 defined by an abrupt change in sediment grain size between the sandy foreshore and the upper Accepted in revised form shoreface that is characterised by frequently occurring large clasts. The main parts of both fine and 19 April 2018 large clasts appear to be locally derived. Seaward-dipping reflections form downlap points, which Published online are clearly identified in all beach-ridge GPR profiles. Most of them are located at the boundary 7 September 2018 between a unit with reflection characteristics representing palaeo-foreshore deposits and a deeper and more complex radar unit characterised by diffractions; the deeper unit is not penetrated to large depths by the GPR signals. Based on observations of the active shoreface regime, large clasts are interpreted to give rise to scattering observed near the top of the deeper radar unit. -
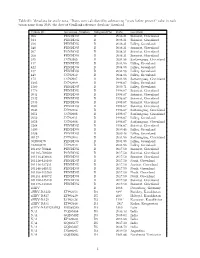
Value in Each Taxon Name from 2016, the Date of Genbank Reference Database Download
Table S1: Metadata for study taxa. ∗Dates were calculated by subtracting "years before present" value in each taxon name from 2016, the date of GenBank reference database download. Taxon ID Accession Number Subgenotype Date Location 208 PENDING D 2008.21 Sisimiut, Greenland 214 PENDING D 2008.21 Sisimiut, Greenland 218 PENDING D 2008.21 Itilleq, Greenland 248 PENDING D 2008.21 Sisimiut, Greenland 267 PENDING D 2008.21 Sisimiut, Greenland 268 PENDING D 2008.21 Sisimiut, Greenland 345 JN792905 D 2004.96 Sarfannguaq, Greenland 417 PENDING D 2004.96 Itilleq, Greenland 422 PENDING D 2004.96 Itilleq, Greenland 437 PENDING D 2004.96 Itilleq, Greenland 449 JN792912 D 2004.96 Itilleq, Greenland 473 JN792907 D 2004.96 Sarfannguaq, Greenland 1205 JN792909 D 1998.87 Itilleq, Greenland 1509 PENDING D 2009.71 Itilleq, Greenland 1776 PENDING D 1998.87 Sisimiut, Greenland 2031 PENDING D 1998.87 Sisimiut, Greenland 2132 PENDING D 1998.87 Sisimiut, Greenland 2335 PENDING D 1998.87 Sisimiut, Greenland 2903 PENDING D 1998.87 Sisimiut, Greenland 2943 JN792904 D 1998.87 Sarfannguaq, Greenland 2951 JN792908 D 1998.87 Sarfannguaq, Greenland 2952 JN792911 D 1998.87 Itilleq, Greenland 2958 JN792906 D 1998.87 Sarfannguaq, Greenland 3288 PENDING D 1998.87 Sisimiut, Greenland 5180 PENDING D 2009.46 Itilleq, Greenland 5198 PENDING D 2009.46 Itilleq, Greenland 30127 JN792903 D 2004.96 Sarfannguaq, Greenland 302000479 PENDING D 2004.96 Itilleq, Greenland 312000479 JN792910 D 2004.96 Itilleq, Greenland 101102-700441 PENDING D 2017.50 Sisimiut, Greenland 101105-706980 -

The Necessity of Close Collaboration 1 2 the Necessity of Close Collaboration the Necessity of Close Collaboration
The Necessity of Close Collaboration 1 2 The Necessity of Close Collaboration The Necessity of Close Collaboration 2017 National Spatial Planning Report 2017 autumn assembly Ministry of Finances and Taxes November 2017 The Necessity of Close Collaboration 3 The Necessity of Close Collaboration 2017 National Spatial Planning Report Ministry of Finances and Taxes Government of Greenland November 2017 Photos: Jason King, page 5 Bent Petersen, page 6, 113 Leiff Josefsen, page 12, 30, 74, 89 Bent Petersen, page 11, 16, 44 Helle Nørregaard, page 19, 34, 48 ,54, 110 Klaus Georg Hansen, page 24, 67, 76 Translation from Danish to English: Tuluttut Translations Paul Cohen [email protected] Layout: allu design Monika Brune www.allu.gl Printing: Nuuk Offset, Nuuk 4 The Necessity of Close Collaboration Contents Foreword . .7 Chapter 1 1.0 Aspects of Economic and Physical Planning . .9 1.1 Construction – Distribution of Public Construction Funds . .10 1.2 Labor Market – Localization of Public Jobs . .25 1.3 Demographics – Examining Migration Patterns and Causes . 35 Chapter 2 2.0 Tools to Secure a Balanced Development . .55 2.1 Community Profiles – Enhancing Comparability . .56 2.2 Sector Planning – Enhancing Coordination, Prioritization and Cooperation . 77 Chapter 3 3.0 Basic Tools to Secure Transparency . .89 3.1 Geodata – for Structure . .90 3.2 Baseline Data – for Systematization . .96 3.3 NunaGIS – for an Overview . .101 Chapter 4 4.0 Summary . 109 Appendixes . 111 The Necessity of Close Collaboration 5 6 The Necessity of Close Collaboration Foreword A well-functioning public adminis- by the Government of Greenland. trative system is a prerequisite for a Hence, the reports serve to enhance modern democratic society. -

Birthplace of Icebergs
WEST GREENLAND: DISKO BAY СRUISE WELCOME TO BIRTHPLACE OF ICEBERGS Though seemingly larger than life, Disko Bay is almost not large enough to hold its many wonders. This voyage centers on Disko Bay, a region on the west coast of Greenland famous for its icebergs, whales, and friendly Inuit villages. It departs from the capital of Greenland city of Nuuk and finishes in Kangerlussuaq. Attractions include volcanic landscapes, museums, iconic churches in numerous settlements, Greenlandic sled dogs, and stunning scenery everywhere. Our area of exploration encompasses a wealth of natural wonders such as the famous Ilulissat Icefjord, the beautiful Eqip Sermia glacier, the heart-shaped Uummannaq mountain, and breathtaking fjords, such as Nuuk fjord, reaching deep into the mountainous wilderness of Greenland. DATES - 22 May; 30 May 2018 DURATION - 9 days EMBARKATION - Nuuk (Greenland) DISEMBARKATION - Kangerlussuaq (Greenland) SHIP - M/V Sea Spirit From $5,695 (Double) landscape contains hot springs, red basalt mountains, HIGHLIGHTS lush greenery, and amazing rock formations. The small, ILULISSAT ICEFJORD traditional town of Qeqertarsuaq is among of The spectacular Ilulissat Icefjord is a UNESCO World Greenland’s oldest. Heritage Site. At the head of this steady stream of ice The waters around Disko Island are home to a variety of lies Sermeq Kujalleq (Jakobshavn Glacier), an outlet of whale species including humpback, fin, minke, and the Greenland Ice Sheet through which around 46 cubic bowhead (also known as the Greenland right whale). kilometers of ice are calved annually into the sea. The abundance of icebergs makes whale watching here Situated nearby is the town of Ilulissat, which offers a particularly awesome experience. -

Årsregnskab 2019
ÅRSREGNSKAB 2019 Transmissionslinje fra Sisimiut vandkraftværk FORORD Et år med medvind og modvind Vi har høje ambitioner i Nukissiorfiit. Der er meget, vi gerne vil udret problemer med vandforsyningen i Uummannaq. Der var behov for en te for samfundet og for virksomheden. Det gælder ikke mindst i for ekstraordinær indsats for at sikre rent drikkevand til Uummannaq og hold til at bidrage til den grønne omstilling og til at fastholde et lavt genetablere den normale vandforsyning. prisniveau for el, vand og varme. Vi arbejder for miljømæssig og øko nomisk bæredygtighed – i Nukissiorfiit og i samfundet. Regnskabet for 2019 viser, at Nukissiorfiits økonomi er lettere udfor dret. Vi havde samlet set et mindre underskud på 5 mio. kr., som dog I 2019 har vi kunnet glæde os over, at vi har nået en række af de mål, kunne have været meget værre. Vi formåede imidlertid at fastholde vi har sat os. Vi har dog også mødt udfordringer, som har betydet, at et lavt prisniveau på el, vand og varme på trods af et stigende pres på vi har været nødt til at tilrettelægge en del af vores arbejde anderle økonomien fra flere sider. des, end vi forventede ved årets start. I 2019 sagde vi farvel til Nukissiorfiits mangeårige energidirektør, Vi er stolte over, at vi er nået langt med planlægningen af, hvordan vi Michael Pedersen. Indtil der udpeges en ny energidirektør, bestrides kan bruge vedvarende energi over hele landet i stedet for olie. Vi er stillingen af Nukissiorfiits økonomidirektør, Claus Andersen-Aagaard. også stolte over, at vores indsats for høj vandkvalitet i bygderne har betydet, at de sidste kogeanbefalinger blev ophævet i 2019. -

The Northwest Passage AUGUST 22 SEPTEMBER 7 & SEPTEMBER 7 23, 2020 2 Contents
EXPEDITION CRUISE The Northwest Passage AUGUST 22SEPTEMBER 7 & SEPTEMBER 723, 2020 2 Contents 5 Itinerary: Into the Northwest Passage 11 Itinerary: Out of the Northwest Passage SHIP 16 The Ocean Endeavour 18 Life Aboard the Ocean Endeavour PRICING, INCENTIVES, & REGISTRATION 20 Berth Prices 21 League of Adventurers & Incentives 22 Program Enhancements 23 Important Information Photography: Courtesy of Newfoundland & Labrador Tourism, Andrew Stewart, Jean Weller, Jason van Bruggen, Mike Beedell, Martin Aldrich, Scott Forsyth, Kristian Bogner, Craig Minielly, Dennis Minty, Andre Gallant, and Vladimir Rajevac 3 4 EXPEDITION CRUISE Into the Northwest Passage AUG.22SEPT. 7, 2020 Starts: Toronto, ON From $10,995 to $25,095 USD Ends: Calgary, AB per person (details pp.20) Aboard the Ocean Endeavour Charter Flights (details pp.20) ITINERARY Day 1: Kangerlussuaq, GL Day 2: Sisimiut Coast BEECHEY Day 3: Ilulissat ISLAND DEVON ISLAND GREENLAND TALLURUTIUP Day 4–5: Western Greenland VICTORIA PRINCE OF IMANGA LANCASTER ISLAND WALES ISLAND SOUND Day 6: At Sea — Davis Strait INLET IQALUKTUUTTIAQ PRINCE REGENT CORONATION (CAMBRIDGE BAY) Day 7: Mittimatalik (Pond Inlet), NU GULF QUEEN MAUD Mittimatalik GULF (Pond Inlet) Tallurutiup Imanga (Lancaster Kugluktuk Day 8–10: Uqsuqtuuq (Coppermine) BAFFIN Ilulissat (Gjoa Haven) ISLAND Sound) & Devon Island Sisimiut NUNAVUT Coast Day 11: Beechey Island Kangerlussuaq Day 12–13: Prince Regent Inlet CANADA DAVIS STRAIT Day 14–16: Kitikmeot Region Day 17: Kugluktuk, NU HIGHLIGHTS • Cross the Arctic Circle as you sail the length • Watch for marine mammals and wildlife in of Sondre Stromfjord—168 kilometres! Tallurutiup Imanga (Lancaster Sound) Marine • Cruise among icebergs at Ilulissat Icefjord, Protected Area a UNESCO World Heritage Site • Seek polar bears, seabirds, and other Arctic • Visit Queen Maud Gulf, home to the wrecks of wildlife in pristine natural environments the Franklin ships, HMS Erebus and HMS Terror Adventure Canada itineraries may be subject to change without notice due to weather, 5 ice, and sea conditions. -

Magnetic Anomalies and Metamorphic Boundaries in the Southern Nagssugtoqidian Orogen, West Greenland
Magnetic anomalies and metamorphic boundaries in the southern Nagssugtoqidian orogen, West Greenland John A. Korstgård, Bo Møller Stensgaard and Thorkild M. Rasmussen Within the southern Nagssugtoqidian orogen in West Greenland metamorphic terrains of both Ar- chaean and Palaeoproterozoic ages occur with metamorphic grade varying from low amphibolite facies to granulite facies. The determination of the relative ages of the different metamorphic terrains is greatly aided by the intrusion of the 2 Ga Kangâmiut dyke swarm along a NNE trend. In Archaean areas dykes cross-cut gneiss structures, and the host gneisses are in amphibolite to granulite facies. Along Itilleq strong shearing in an E–W-oriented zone caused retrogression of surrounding gneisses to low amphibolite facies. Within this Itivdleq shear zone Kangâmiut dykes follow the E–W shear fab- rics giving the impression that dykes were reoriented by the shearing. However, the dykes remain largely undeformed and unmetamorphosed, indicating that the shear zone was established prior to dyke emplacement and that the orientation of the dykes here was governed by the shear fabric. Me- tamorphism and deformation north of Itilleq involve both dykes and host gneisses, and the metamor- phic grade is amphibolite facies increasing to granulite facies at the northern boundary of the south- ern Nagssugtoqidian orogen. Here a zone of strong deformation, the Ikertôq thrust zone, coincides roughly with the amphibolite–granulite facies transition. Total magnetic field intensity anomalies from aeromagnetic data coincide spectacularly with metamorphic boundaries and reflect changes in content of the magnetic minerals at facies transitions. Even the nature of facies transitions is apparent. Static metamorphic boundaries are gradual whereas dynamic boundaries along deformation zones are abrupt. -

Hydrological and Meteorological Investigations in a Periglacial Lake Catchment Near Kangerlussuaq, West Greenland – Presentation of a New Multi-Parameter Data Set
Earth Syst. Sci. Data, 7, 93–108, 2015 www.earth-syst-sci-data.net/7/93/2015/ doi:10.5194/essd-7-93-2015 © Author(s) 2015. CC Attribution 3.0 License. Hydrological and meteorological investigations in a periglacial lake catchment near Kangerlussuaq, west Greenland – presentation of a new multi-parameter data set E. Johansson1,2, S. Berglund4, T. Lindborg1,3, J. Petrone2, D. van As5, L.-G. Gustafsson6, J.-O. Näslund1,2, and H. Laudon3 1Department of Physical Geography, Bert Bolin Centre for Climate Research, Stockholm University, 106 91 Stockholm, Sweden 2Swedish Nuclear Fuel and Waste Management Co, Box 250, 101 24 Stockholm, Sweden 3Department of forest ecology and management, Swedish University of Agricultural Science, 901 83 Umeå, Sweden 4Hydroresearch AB, St. Marknadsvägen 15S (12th floor), 183 34 Täby, Sweden 5Geological Survey of Denmark and Greenland (GEUS), Øster Voldgade 10, 1350 Copenhagen, Denmark 6DHI Sweden AB, Honnörsgatan 16, Box 3287, 350 53 Växjö, Sweden Correspondence to: E. Johansson ([email protected]) Received: 15 October 2014 – Published in Earth Syst. Sci. Data Discuss.: 16 December 2014 Revised: 20 April 2015 – Accepted: 6 May 2015 – Published: 29 May 2015 Abstract. Few hydrological studies have been conducted in Greenland, other than on glacial hydrology associ- ated with the ice sheet. Understanding permafrost hydrology and hydroclimatic change and variability, however, provides key information for understanding climate change effects and feedbacks in the Arctic landscape. This paper presents a new, extensive, and detailed hydrological and meteorological open access data set, with high temporal resolution from a 1.56 km2 permafrost catchment, with a lake underlain by a through-talik close to the ice sheet in the Kangerlussuaq region, western Greenland. -

Danish Lithosphere Centre Studies of the Nagssugtoqidian Orogen,West Greenland
Archaean and Palaeoproterozoic orogenic processes: Danish Lithosphere Centre studies of the Nagssugtoqidian orogen,West Greenland Flemming Mengel, Jeroen A. M. van Gool, Eirik Krogstad and the 1997 field crew The Danish Lithosphere Centre (DLC) was established by Escher et al. (1970, 1975), and was viewed as a in 1994 and one of its principal objectives in the first frontal thrust, across which the southern Nagssugtoqidian five-year funding cycle is the study of Precambrian oro- orogen was translated towards the southern foreland. genic processes. This work initially focused on the ther- Figure 1 shows the main lithotectonic units of the mal and tectonic evolution of the Nagssugtoqidian Nagssugtoqidian orogen (from Marker et al. 1995; van orogen of West Greenland. Gool et al. 1996). The southern foreland consists of During the first two field seasons (1994 and 1995) Archaean granulite facies tonalitic-granodioritic gneisses most efforts were concentrated in the southern and cut by mafic dykes of the Palaeoproterozoic Kangâmiut central portions of the orogen. The 1997 field season swarm. In the SNO (southern Nagssugtoqidian orogen) was the third and final in the project in the Nagssug- gneisses and dykes from the foreland were deformed toqidian orogen and emphasis was placed on the cen- and metamorphosed in amphibolite facies and trans- tral and northern parts of the orogen in order to complete posed into NNW-dipping structures during SSE-directed the lithostructural study of the inner Nordre Strømfjord ductile thrusting. Towards the contact between the SNO area and to investigate the northern margin of the oro- and CNO (central Nagssugtoqidian orogen), the metamor- gen (NNO in Fig. -

Download Free
ENERGY IN THE WEST NORDICS AND THE ARCTIC CASE STUDIES Energy in the West Nordics and the Arctic Case Studies Jakob Nymann Rud, Morten Hørmann, Vibeke Hammervold, Ragnar Ásmundsson, Ivo Georgiev, Gillian Dyer, Simon Brøndum Andersen, Jes Erik Jessen, Pia Kvorning and Meta Reimer Brødsted TemaNord 2018:539 Energy in the West Nordics and the Arctic Case Studies Jakob Nymann Rud, Morten Hørmann, Vibeke Hammervold, Ragnar Ásmundsson, Ivo Georgiev, Gillian Dyer, Simon Brøndum Andersen, Jes Erik Jessen, Pia Kvorning and Meta Reimer Brødsted ISBN 978-92-893-5703-6 (PRINT) ISBN 978-92-893-5704-3 (PDF) ISBN 978-92-893-5705-0 (EPUB) http://dx.doi.org/10.6027/TN2018-539 TemaNord 2018:539 ISSN 0908-6692 Standard: PDF/UA-1 ISO 14289-1 © Nordic Council of Ministers 2018 Cover photo: Mats Bjerde Print: Rosendahls Printed in Denmark Disclaimer This publication was funded by the Nordic Council of Ministers. However, the content does not necessarily reflect the Nordic Council of Ministers’ views, opinions, attitudes or recommendations. Rights and permissions This work is made available under the Creative Commons Attribution 4.0 International license (CC BY 4.0) https://creativecommons.org/licenses/by/4.0 Translations: If you translate this work, please include the following disclaimer: This translation was not produced by the Nordic Council of Ministers and should not be construed as official. The Nordic Council of Ministers cannot be held responsible for the translation or any errors in it. Adaptations: If you adapt this work, please include the following disclaimer along with the attribution: This is an adaptation of an original work by the Nordic Council of Ministers. -

Pdf Dokument
Udskriftsdato: 27. september 2021 BEK nr 1785 af 24/11/2020 (Gældende) Bekendtgørelse om ændring af den fortegnelse over valgkredse, der indeholdes i lov om folketingsvalg i Grønland Ministerium: Social og Indenrigsministeriet Journalnummer: Social og Indenrigsmin., j.nr. 20203732 Bekendtgørelse om ændring af den fortegnelse over valgkredse, der indeholdes i lov om folketingsvalg i Grønland I medfør af § 8, stk. 1, i lov om folketingsvalg i Grønland, jf. lovbekendtgørelse nr. 916 af 28. juni 2018, som ændret ved bekendtgørelse nr. 584 af 3. maj 2020, fastsættes: § 1. Fortegnelsen over valgkredse i Grønland affattes som angivet i bilag 1 til denne bekendtgørelse. § 2. Bekendtgørelsen træder i kraft den 5. december 2020. Social- og Indenrigsministeriet, den 24. november 2020 Nikolaj Stenfalk / Christine Boeskov BEK nr 1785 af 24/11/2020 1 Bilag 1 Ilanngussaq Fortegnelse over valgkredse i hver kommune Kommuneni tamani qinersivinnut nalunaarsuut Kommune Valgkredse i Valgstedet eller Valgkredsens område hver kommune afstemningsdistrikt (Tilknyttede bosteder) (Valgdistrikt) (Afstemningssted) Kommune Nanortalik 1 Nanortalik Nanortalik Kujalleq 2 Aappilattoq (Kuj) Aappilattoq (Kuj) Ikerasassuaq 3 Narsarmijit Narsarmijit 4 Tasiusaq (Kuj) Tasiusaq (Kuj) Nuugaarsuk Saputit Saputit Tasia 5 Ammassivik Ammassivik Qallimiut Qorlortorsuaq 6 Alluitsup Paa Alluitsup Paa Alluitsoq Qaqortoq 1 Qaqortoq Qaqortoq Kingittoq Eqaluit Akia Kangerluarsorujuk Qanisartuut Tasiluk Tasilikulooq Saqqaa Upernaviarsuk Illorsuit Qaqortukulooq BEK nr 1785 af 24/11/2020