The Regulation of Mitochondrial Uncoupling Proteins in the Heart Karl Robert Laskowski Yale University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Uncoupling Protein 3 in Human Physiology
The role of uncoupling protein 3 in human physiology W. Timothy Garvey J Clin Invest. 2003;111(4):438-441. https://doi.org/10.1172/JCI17835. Commentary Obesity is simply understood as an imbalance between energy intake and expenditure in favor of weight accretion. However, the human biological interface between food consumption and energy dissipation results in broad individual differences in eating behavior, physical activity, and efficiency of fuel storage and metabolism. In particular, the basal metabolic rate, which accounts for the greatest portion of overall energy expenditure, can vary almost twofold among individuals. Classically, three major biochemical systems are believed to contribute to basal thermogenesis: futile cycles, Na+/K+ATPase activity, and mitochondrial proton leak. The latter is the most important quantitative contributor and can explain up to 50% of the basal metabolic rate (1). The molecular basis of mitochondrial proton leak is unclear, despite its importance in the understanding of energy balance and its potential as a therapeutic target for obesity treatment. The article by Hesselink and colleagues in this issue of the JCI (2) addresses whether uncoupling protein 3 contributes to mitochondrial proton leak in human skeletal muscle. Mitochondrial respiration and oxidative phosphorylation The oxidation of fatty acids and pyruvate takes place in mitochondria, where energy is converted into ATP for use in cellular processes. Reducing equivalents are extracted from substrates and sequentially passed from electron donors (reductants) to acceptors (oxidants) along the mitochondrial respiratory chain to molecular oxygen. The electron transport system is located on […] Find the latest version: https://jci.me/17835/pdf COMMENTARY See the related article beginning on page 479. -

Perspectives in Diabetes Uncoupling Proteins 2 and 3 Potential Regulators of Mitochondrial Energy Metabolism Olivier Boss, Thilo Hagen, and Bradford B
Perspectives in Diabetes Uncoupling Proteins 2 and 3 Potential Regulators of Mitochondrial Energy Metabolism Olivier Boss, Thilo Hagen, and Bradford B. Lowell Mitochondria use energy derived from fuel combustion fuels and oxygen are converted into carbon dioxide, water, to create a proton electrochemical gradient across the and ATP (Fig. 1). The key challenge for the organism is to reg- mitochondrial inner membrane. This intermediate form ulate these many steps so that rates of ATP production are of energy is then used by ATP synthase to synthesize equal to rates of ATP utilization. This is not a small task given AT P. Uncoupling protein-1 (UCP1) is a brown fat–spe- that rates of ATP utilization can quickly increase severalfold cific mitochondrial inner membrane protein with proton (up to 100-fold in muscle during contraction). transport activity. UCP1 catalyzes a highly regulated proton leak, converting energy stored within the mito- Fuel metabolism and oxidative phosphorylation consist chondrial proton electrochemical potential gradient to of many tightly coupled enzymatic reactions (Fig. 1), which heat. This uncouples fuel oxidation from conversion of are regulated, in part, by ADP availability. Control by ADP is ADP to AT P. In rodents, UCP1 activity and brown fat accounted for by the chemiosmotic hypothesis of Mitchell (1). contribute importantly to whole-body energy expendi- Oxidation of fuels via the electron transport chain generates ture. Recently, two additional mitochondrial carriers a proton electrochemical potential gradient ( µH+) across with high similarity to UCP1 were molecularly cloned. the mitochondrial inner membrane. Protons reenter the In contrast to UCP1, UCP2 is expressed widely, and mitochondrial matrix via ATP synthase (F0F1-A TPase) in a UCP3 is expressed preferentially in skeletal muscle. -

Uncoupling Protein-3 (UCP3): a Mitochondrial Carrier in Search of a Function
International Journal of Obesity (1999) 23, Suppl 6, S43±S45 ß 1999 Stockton Press All rights reserved 0307±0565/99 $12.00 http://www.stockton-press.co.uk/ijo Uncoupling protein-3 (UCP3): A mitochondrial carrier in search of a function BB Lowell1* 1Division of Endocrinology, Department of Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts, USA UCP3 is a mitochondrial protein with high homology to the established uncoupling protein, UCP1. Its high degree of homology to UCP1 suggests that UCP3 may be a true uncoupling protein. Preliminary biochemical studies are consistent with UCP3 having uncoupling activity. However, detailed functional studies are required to understand the true biochemical and physiological purpose of UCP3. These efforts should be aided by identi®cation of humans with inactivating mutations and=or the generation of gene knockout mice lacking UCP3. Keywords: brown adipose tissue; skeletal muscle; adipose tissue; UCP3 isoforms Introduction ( 71% identical at the amino acid level). UCP3 is much less homologous to other members of the mitochondrial carrier super family: oxoglutarate car- Uncoupling protein-1 (UCP1) is a brown fat-speci®c, rier ( 32% identical), citrate carrier ( 25% identi- mitochondrial inner membrane protein with proton cal), carnitine carrier ( 25% identical), phosphate transport activity. This activity uncouples fuel con- carrier ( < 25% identical) and ADP=ATP carrier sumption from the conversion of ADP to ATP, ( < 25% identical). Based upon the high homology thereby releasing stored energy as heat. In rodents, of UCP3 with UCP1, compared to its lower homology UCP1 activity and brown fat contribute importantly to with other members of the mitochondrial carrier whole body energy expenditure. -

The Uncoupling Protein Homologues: UCP1, UCP2, UCP3, Stucp
Biochem. J. (2000) 345, 161–179 (Printed in Great Britain) 161 REVIEW ARTICLE The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP Daniel RICQUIER1 and Fre! de! ric BOUILLAUD Centre de Recherche sur l’Endocrinologie Mole! culaire et le De! veloppement (CEREMOD), Centre National de la recherche Scientifique (CNRS – Unit 9078), 9 rue Jules Hetzel, 92190 Meudon, France Animal and plant uncoupling protein (UCP) homologues form a energy expenditure in humans. The UCPs may also be involved subfamily of mitochondrial carriers that are evolutionarily re- in adaptation of cellular metabolism to an excessive supply of lated and possibly derived from a proton}anion transporter substrates in order to regulate the ATP level, the NAD+}NADH ancestor. The brown adipose tissue (BAT) UCP1 has a marked ratio and various metabolic pathways, and to contain superoxide and strongly regulated uncoupling activity, essential to the production. A major goal will be the analysis of mice that either maintenance of body temperature in small mammals. UCP lack the UCP2 or UCP3 gene or overexpress these genes. Other homologues identified in plants are induced in a cold environment aims will be to investigate the possible roles of UCP2 and UCP3 and may be involved in resistance to chilling. The biochemical in response to oxidative stress, lipid peroxidation, inflammatory activities and biological functions of the recently identified processes, fever and regulation of temperature in certain specific mammalian UCP2 and UCP3 are not well known. However, parts of the body. recent data support a role for these UCPs in State 4 respiration, respiration uncoupling and proton leaks in mitochondria. -

Mitochondrial Uncoupling Protein 2 (UCP2), but Not UCP3, Is Sensitive to Oxygen Concentration in Cells
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.01.181768; this version posted July 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Article Mitochondrial uncoupling protein 2 (UCP2), but not UCP3, is sensitive to oxygen concentration in cells Karolina E. Hilse1, Anne Rupprecht1,2, Kristopher Ford1, Olena Andrukhova1,†, Reinhold Erben and Elena E. Pohl1,* 1 Institute of Physiology, Pathophysiology and Biophysics, Department of Biomedical Sciences, University of Veterinary Medicine, Vienna, Austria 2 Institute of Pharmacology and Toxicology, Rostock University Medical Center, Germany * Correspondence: [email protected] † Deceased 3 October 2017 Received: date; Accepted: date; Published: date Abstract: One of the important hallmarks of cardiovascular disease is mitochondrial dysfunction, which results in abnormal energy metabolism and increased ROS production in cardiomyocytes. Members of the mitochondrial uncoupling protein family, UCP2 and UCP3, are thought to be beneficial by reducing ROS due to mild uncoupling. More recent hypotheses suggest the involvement of both proteins in cell metabolism by the transport of yet unknown substrates. The protein expression pattern under physiological and pathological conditions is an important clue for the evaluation of UCP2/UCP3 function, however, there is still no consensus about it. Previously, we demonstrated that only UCP3 is present in the adult murine heart under physiological conditions and correlated it with the predominant use of fatty acids for oxidation. In contrast, UCP2 was found only in very young (stem cell - like) cardiomyocytes, that rely mostly on glycolysis. -

(UCP2) and Uncoupling Protein-3 (UCP3) Expression in Adipose Tissue and Skeletal Muscle in Humans
International Journal of Obesity (1999) 23, Suppl 6, S64±S67 ß 1999 Stockton Press All rights reserved 0307±0565/99 $12.00 http://www.stockton-press.co.uk/ijo Uncoupling protein-2 (UCP2) and uncoupling protein-3 (UCP3) expression in adipose tissue and skeletal muscle in humans D Langin1*, D Larrouy1, P Barbe1, L Millet1, N Viguerie-Bascands1, F Andreelli2, M Laville2 and H Vidal2 1Unite INSERM 317, Institut Louis Bugnard, Universite Paul Sabatier, HoÃpital Rangueil, Toulouse, France; and 2Unite INSERM 449 et Centre de Recherche en Nutrition Humaine de Lyon, Faculte de MeÂdecine LaeÈnnec, Lyon, France Uncoupling protein-2 (UCP2) and uncoupling protein-3 (UCP3) are mitochondrial proteins that may play a role in the control of energy expenditure by uncoupling respiration from ATP synthesis. The present review focuses on data obtained in humans. UCP2 is widely expressed in the body, whereas UCP3 expression is restricted to skeletal muscle. Positive correlations have been reported between UCP2 mRNA concentrations in adipose tissue, UCP3 mRNA concentrations in skeletal muscle, and components of the metabolic rate. Fasting induces an up-regulation of UCP2 and UCP3 mRNA expression. In vivo and in vitro studies suggest that fatty acids could modulate uncoupling protein gene expression. The putative relationship between obesity, energy expenditure and uncoupling protein expression, and the unexpected rise in UCP2 and UCP3 mRNA concentrations during short-term fasting, are discussed in view of the recent data obtained in rodents and cell lines. Keywords: energy expenditure; fatty acid; adipose tissue; skeletal muscle; obesity; Uncoupling protein-2 (UCP2) and UCP3 mRNAs are markedly different. -
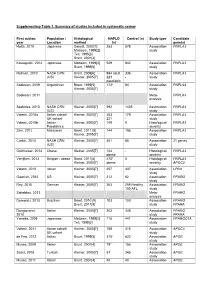
Supplementary Table 1. Summary of Studies Included in Systematic Review
Supplementary Table 1. Summary of studies included in systematic review First author, Population / Histological NAFLD Control (n) Study type Candidate year Location method (n) gene(s) Hotta, 2010 Japanese Sanyal, 2002[1] 253 578 Association PNPLA3 Matteoni, 1999[2] study Teli, 1995[3] Brunt, 2001[4] Kawaguchi, 2012 Japanese Matteoni, 1999[2] 529 942 Association PNPLA3 Brunt, 1999[5] study Rotman, 2010 NASH CRN Brunt, 2009[6] 894 adult 336 Association PNPLA3 (US) Kleiner, 2005[7] 223 - study paediatric Sookoian, 2009 Argentinian Brunt, 1999[5] 172* 94 Association PNPLA3 Kleiner, 2005[7] study Sookoian, 2011 Meta- PNPLA3 analysis Speliotes, 2010 NASH CRN Kleiner, 2005[7] 592 1405 Association PNPLA3 (US) study Valenti, 2010a Italian cohort/ Kleiner, 2005[7] 253 179 Association PNPLA3 UK cohort 321 - study Valenti, 2010b Italian - Kleiner, 2005[7] 149 0 Histological PNPLA3 Paediatrics severity Zain, 2012 Malaysian Brunt, 2011 [8] 144 198 Association PNPLA3 Kleiner, 2005[7] study Corbin, 2013 NASH CRN Kleiner, 2005[7] 361 85 Association 21 genes (US) study Guichelaar, 2013 Obese Kleiner, 2005[7] 144 - Histological PNPLA3 obese severity Verrijken, 2013 Belgian - obese Brunt, 2011[8] 470* 0 Histological PNPLA3 Kleiner, 2005[7] obese severity APOC3 Valenti, 2012 Italian Kleiner, 2005[7] 257 337 Association LPIN1 study Gawrieh, 2012 US Kleiner, 2005[7] 212 62 Association PPARG study Rey, 2010 German Kleiner, 2005[7] 263 259 Healthy Association PPARG 100 AFL study Sahebkar, 2013 Meta- PPARG analysis Domenici, 2013 Brazilian Brunt, 2010 [9] 103 103 -

UCP1, UCP2 and UCP3: Are They True Uncouplers of Respiration?
International Journal of Obesity (1999) 23, Suppl 6, S19±S23 ß 1999 Stockton Press All rights reserved 0307±0565/99 $12.00 http://www.stockton-press.co.uk/ijo UCP1, UCP2 and UCP3: Are they true uncouplers of respiration? F Bouillaud1* 1CEREMOD, CNRS, Meudon, France The thermogenesis in brown adipose tissue (BAT) is due to the activity of a mitochondrial uncoupling protein (UCP1). This protein allows the protons pumped by the respiratory chain to re-enter the matrix without ATP synthesis. Therefore respiration is dramatically increased and produces only heat. The discovery of genes showing strong similarities with the UCP1 gene and expressed in other tissues raised the possibility that these proteins participate in the proton leak observed in mitochondria, and therefore participate in the regulation of energy expenditure. The recombinant expression of UCP1, UCP2 and UCP3 in yeast allows the comparison of the coupling state of yeast mitochondria in the presence or absence of these proteins. Keywords: mitochondria; proton leak; yeast; Saccharomyces cerevisiae Introduction inhibit or mask naturally present uncoupling mechan- isms of physiological relevance. An example is BAT mitochondria, where the uncoupling protein UCP1 is The ®rst hypothesis proposed with discovery of responsible for heat production. sequences highly similar to the brown adipose tissue (BAT) uncoupling protein (UCP1),1±4 was that these proteins had a similar activity. In this paper we will re-examine several results obtained with UCP1 that are relevant here, and will present -

UCP3 Gene Is Associated with Body Mass Index in Humans in Interaction with Physical Activity
Diabetologia (2000) 43: 245±249 Ó Springer-Verlag 2000 A genetic variation in the 5 ¢ flanking region of the UCP3 gene is associated with body mass index in humans in interaction with physical activity S.Otabe1, K.Clement2, C.Dina1, V.Pelloux2, B.Guy-Grand2, P.Froguel1, F.Vasseur1 1 CNRS UPRESA 8090, Institute of Biology of Lille, CHRU Lille, France 2 Department of Nutrition Hotel-Dieu, Paris, France Abstract BMI in the obese group (p = 0.0031): BMI was higher in TT than in CC or CT patients. Likewise control Aims/hypothesis. In obese French Caucasian subjects subjects with a TT genotype had a higher BMI we previously described a silent UCP3 Tyr99Tyr mu- (p = 0.03). In the obese group, homozygocity for this tation, associated with body mass index. We hypothe- variant is a risk factor for high BMI (odds ratio: 1:75, sised that an unknown polymorphism in the vicinity p = 0.02). Obese patients were divided into tertiles of the gene could contribute to obesity. according to physical activity. In the group with a Methods. Morbidly obese subjects were screened for wild C/C genotype, BMI was negatively associated mutationsin1kbupstreamfromthe UCP3gene.Asso- with physical activity (p = 0.015). ciation studies were done between a variant and obesi- Conclusion/interpretation. The C®T polymorphism ty in 401 morbidly obese and 231 control subjects. in the 5 ¢ sequences of the UCP3 gene might contrib- Results. We detected three rare genetic variants and ute to the corpulence in obese and normal weight one polymorphism: a + 5 G®A in exon 1, a ±155 subjects and alter the benefit of physical activity. -

Insulin Restores UCP3 Activity and Decreases Energy Surfeit to Alleviate Lipotoxicity in Skeletal Muscle
2000 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 40: 2000-2010, 2017 Insulin restores UCP3 activity and decreases energy surfeit to alleviate lipotoxicity in skeletal muscle WENJUAN TANG, SUNYINYAN TANG, HONGDONG WANG, ZHIJUAN GE, DALONG ZHU and YAN BI Department of Endocrinology, Drum Tower Hospital Affiliated to Nanjing University Medical School, Nanjing, Jiangsu 210008, P.R. China Received March 12, 2017; Accepted September 19, 2017 DOI: 10.3892/ijmm.2017.3169 Abstract. An early insulin regimen ameliorates glucotox- UCP3 was blocked by the proteasome inhibitor MG132, and icity but also lipotoxicity in type 2 diabetes; however, the insulin reduced UCP3 ubiquitination, thereby prohibiting its underlying mechanism remains elusive. In the present study, degradation. Our findings, focusing on energy balance, provide we investigated the role of mitochondria in lipid regulation a mechanistic understanding of the promising effect of early following early insulin administration in insulin-resistant insulin initiation on lipotoxicity. Insulin, by recovering UCP3 skeletal muscle cells. Male C57BL/6 mice, fed a high-fat diet activity, alleviated energy surfeit and potentiated AMPK- (HFD) for 8 weeks, were treated with insulin for 3 weeks, mediated lipid homeostasis in skeletal muscle cells following and L6 myotubes cultured with palmitate (PA) for 24 h were exposure to PA and in gastrocnemius of mice fed HFD. incubated with insulin for another 12 h. The results showed that insulin facilitated systemic glucose disposal and attenu- Introduction ated muscular triglyceride accumulation in vivo. Recovery of AMP-activated protein kinase (AMPK) phosphorylation, In tandem with the global obesity pandemic, type 2 diabetes inhibition of sterol-regulated element binding protein-1c mellitus (T2DM) has become a major public health issue (1). -

Genotyping for Response to Physical Training
GENOTYPING FOR RESPONSE TO PHYSICAL TRAINING A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By STACY SIMMONS B.S., Wright State University, 2014 _________________________________________________________ 2019 Wright State University WRIGHT STATE UNIVERSITY GRADUATE SCHOOL July 29, 2019 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY Stacy Simmons ENTITLED Genotyping for Response to Physical Training BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science. ___________________________________ Michael Markey, Ph.D. Thesis Director ____________________________________ Madhavi P. Kadakia, Ph.D. Committee on Chair, Department of Biochemistry Final Examination and Molecular Biology _____________________________ Michael Markey, Ph.D. _____________________________ Richard Chapleau, Ph.D. _____________________________ Madhavi Kadakia, Ph.D. _____________________________ Barry Milligan, Ph.D. Interim Dean of the Graduate School ABSTRACT Simmons, Stacy. M.S. Department of Biochemistry and Molecular Biology, Wright State University 2019. Genotyping for Response to Physical Training Understanding the inter-individual variability in physical fitness performance has been the focus of scientific research for decades especially in the United States military. Injury and physical inadequacy cost the U.S military millions of dollars every year. The project PHITE (Precision High Intensity Training through Epigenetics) was funded to investigate this personal complex trait by combing the genetic and epigenetic (non-shared environmental factors) contributions into a single model for physical training response. This project is set up as having 150 male and female recruits between the ages of 18-27 years old. Each participant is randomly put blind into either a high intensity or moderate intensity (group A or group B) 12-week training program. -

Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared Amongst Δγ T, Innate Lymphoid, and Th Cells
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 δγ is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2016; 197:1460-1470; Prepublished online 6 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600643 http://www.jimmunol.org/content/197/4/1460 Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst T, Innate Lymphoid, and Th Cells You Jeong Lee, Gabriel J. Starrett, Seungeun Thera Lee, Rendong Yang, Christine M. Henzler, Stephen C. Jameson and Kristin A. Hogquist J Immunol cites 41 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2016/07/06/jimmunol.160064 3.DCSupplemental This article http://www.jimmunol.org/content/197/4/1460.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. The Journal of Immunology Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst gd T, Innate Lymphoid, and Th Cells You Jeong Lee,* Gabriel J.