A Radionuclide Study of the Clyde Sea Area Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
View Site Report
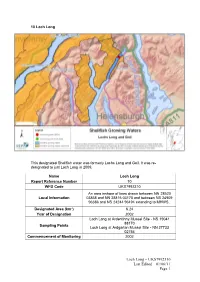
10 Loch Long This designated Shellfish water was formerly Lochs Long and Goil. It was re- designated to just Loch Long in 2009. Name Loch Long Report Reference Number 10 WFD Code UKS7992310 An area inshore of lines drawn between NN 28520 Local Information 03858 and NN 28816 03178 and between NS 24909 96386 and NS 24244 96494 extending to MHWS.. Designated Area (km²) 6.24 Year of Designation 2002 Loch Long at Ardentinny Mussel Site - NS 19041 88170 Sampling Points Loch Long at Ardgartan Mussel Site - NN 27733 02754 Commencement of Monitoring 2002 Loch Long – UKS7992310 Last Edited – 01/06/11 Page 1 10.1 Commercial Shellfish Interests No Crown Estates Commission (CEC) leases have been granted, although there are some natural populations of mussels in the area. The production area has been declassified by the Food Standards Agency due to lack of commercial harvesting. For more information on Food Standards Agency Classification please visit: http://www.food.gov.uk/scotland/safetyhygienescot/shellmonitorscot/shellclassesscot/ 10.2 Bathymetric Information Loch Long has a total length of 26.9 km and a catchment area of 166 km². Two sills divide Loch Long into two water areas or basins. One is located at the loch entrance (near Blairmore) and the other just northwest of Portincaple. The basins have a maximum water depth of 60 and 92 m. Maximum water depth for the loch is 97 m. As a whole, the loch takes on average 6 days to flush, although each basin will have its own flushing characteristics. Fresh/tidal flow ratio which reflects the influence of fresh water on the loch is 0.2, which is low for the size and volume of this loch, implying generally high salinity. -

Newly Renovated Cottage with Views of Loch Goil Ashburn Lochgoilhead, Cairndow, Argyll, Pa24 8Aq
NEWLY RENOVATED COTTAGE WITH VIEWS OF LOCH GOIL ashburn lochgoilhead, cairndow, argyll, pa24 8aq NEWLY RENOVATED COTTAGE WITH VIEWS OF LOCH GOIL Ashburn, Lochgoilhead, Cairndow, Argyll, PA24 8AQ Sun room w Sitting room with dining area w Kitchen w Utility room w Two double bedrooms Shower room w Gardens w Log cabin with music room/office and WC w Private mooring Glasgow Airport: 45 miles, Glasgow city centre: 51 miles Situation Ashburn is situated in the centre of the sought after village of Lochgoilhead in Argyll and is located within the Loch Lomond and Trossachs National Park. Lochgoilhead sits at the northern end of Loch Goil, where life centres around the village shop, village pub and church. The area offers a spectacular landscape of mountains and lochs and is therefore an ideal base for outdoor enthusiasts. There is superb hill walking and climbing as well as excellent fishing and sailing on the loch. The drive to Lochgoilhead from Glasgow passing Loch Lomond, the Arrochar Alps and the Rest and Be Thankful, from where the single track road leads to Lochgoilhead, is a stunning journey. Description Ashburn is a charming detached cottage situated in the heart of the village, on the edge of Loch Goil. The property has been sympathetically refurbished to create a spacious home with modern fixtures and fittings complementing existing period features. The accommodation is all on one level and comprises: sun room with stunning views over the gardens and Loch Goil; sitting room with exposed brick wall, multi fuel stove and dining area; modern fitted kitchen with dining table; utility room; two double bedrooms and a modern fitted shower room. -

Ctog) Target ‘H’ Template for Developing Local Target Setting for Rural Services Priority Areas (Rspas) to Be Completed for Each RSPA
Closing the Opportunity Gap (CtOG) Target ‘h’ Template for Developing Local Target Setting for Rural Services Priority Areas (RSPAs) To be completed for each RSPA CtOG Target ‘h’: By 2008, improve service delivery in rural areas so that agreed improvements to accessibility and quality are achieved for key services in remote and disadvantaged communities. 1. Geography of Rural Service Priority Area Community Planning Partnership Argyll and Bute Datazone number SO1000800 Name of ward within which datazone lies East Lochfyne, Holy Loch Intermediate geography number and name SO2000146, Cowal North 2. Description of services to be targeted for improvement, the boundary and equality impacts 2a) Describe the proposed service(s) to be included in the target. Include generic type (e.g. health/education); service deliver(s); and population to be targeted (e.g. age group) Advice Network online referrals – a collaboration between public, private and voluntary sector agencies to better coordinate provision of advice services. The system allows an organisation to refer a client to the appropriate source of advice that may be located at a distant site and track the referral through to its resolution or closure. This will improve access to advice services and referrals will ensure that the network plays to the strength of its members, thereby improving quality. Community transport – extension of the Cowal Deserve community transport scheme to the datazone area and support for purchase of a community bus to be owned by the Loch Goil Trust. Access to health services and a range of community activities will significantly improve. Extension of the Healthy Communities Collaborative – provision of training, support and materials to a local network that focuses on falls prevention in the elderly. -

Corran Corran Bay, Loch Goil, Argyll and Bute
CORRAN CORRAN BAY, LOCH GOIL, ARGYLL AND BUTE CORRAN, CORRAN BAY, LOCH GOIL, ARGYLL AND BUTE A rare development opportunity to acquire two delightful and secluded properties on Loch Goil within the Loch Lomond & Trossachs National Park Carrick Castle (by boat) 1 mile ■ Lochgoilhead 7 miles (by road) Glasgow 47 miles For sale as a whole Acreage approximately 1.23 acres (0.50 hectares) ■ Stunning shoreline location with wonderful views across Loch Goil ■ Principal house and separate former cottage ■ Total privacy and seclusion ■ Unique development opportunity ■ Easy access by boat ■ Option for alternative access (by foot) ■ Off grid living ■ Within easy reach of Glasgow and airport Stirling Nicky Archibald 01786 434600 07708 322929 [email protected] [email protected] SITUATION DESCRIPTION Corran, comprising Corran House and Corran Cottage, Corran House and Corran Cottage are being offered enjoys a unique positon on the shores of Corran Bay for sale as a whole for the first time in nearly a century at the entrance to Loch Goil. The property, which is and provide the opportunity to create and develop accessible by boat, is approximately 7 miles from the an exceptional, private oasis suitable for permanent, village of Lochgoilhead on the Cowl Peninsula and is holiday or business use (subject to planning). Both situated within The Loch Lomond and The Trossachs properties are detached and surrounded by a National Park. Corran benefits from an elevated, south delightful, mature garden backing onto the extensive facing posiiton and exceptional seclusion and privacy. Argyll Forest Park. Bordering the garden to the front The properties enjoy uninterrupted views across Loch and extending the length of the site, is a substantial Goil with superb panoramic mountain vistas providing stone sea-wall providing excellent protection. -

The Special Landscape Qualities of the Loch Lomond and the Trossachs National Park
COMMISSIONED REPORT Commissioned Report No. 376 The Special Landscape Qualities of the Loch Lomond and The Trossachs National Park (iBids and Project no 648) Produced in partnership with The Loch Lomond and the Trossachs National Park Authority For further information on this report please contact: Dr James Fenton Scottish Natural Heritage Great Glen House INVERNESS IV3 8NW Telephone: 01463-725 000 E-mail: [email protected] This report should be quoted as: Scottish Natural Heritage and Loch Lomond and The Trossachs National Park Authority (2010). The special landscape qualities of the Loch Lomond and The Trossachs National Park. Scottish Natural Heritage Commissioned Report, No.376 (iBids and Project no 648). This report, or any part of it, should not be reproduced without the permission of Scottish Natural Heritage. This permission will not be withheld unreasonably. © Scottish Natural Heritage 2010 COMMISSIONED REPORT Summary The Special Landscape Qualities of the Loch Lomond and The Trossachs National Park Commissioned Report No. 376 (iBids and Project no 648) Contractor: SNH project staff Year of publication: 2010 Background In 2007/8 Scottish Natural Heritage used a standard method to determine the special qualities of Scotland’s National Scenic Areas. In 2009, in partnership with the National Park Authorities, this work was extended to determine the special qualities of the two National Parks, including the National Scenic Areas within them. The result of the work for the Loch Lomond and the Trossachs National Park is reported here. Main findings • The park is large and diverse with significantly different landscape characteristics in different areas. Hence as well as listing the landscape qualities which are generic across the park, the qualities for the four landscape areas of Argyll Forest, Loch Lomond, Breadalbane and the Trossachs are also given. -

Newly Renovated Family Villa with Views of Loch Goil Heathbank Lochgoilhead, Cairndow, Argyll, Pa24 8Ah
NEWLY RENOVATED FAMILY VILLA WITH VIEWS OF LOCH GOIL heathbank lochgoilhead, cairndow, argyll, pa24 8ah NEWLY RENOVATED FAMILY VILLA WITH VIEWS OF LOCH GOIL heathbank lochgoilhead, cairndow, argyll, pa24 8ah Sitting room with dining area w kitchen/family room w utility room w wc w gallery landing w 3 double bedrooms w additional single bedroom w family bathroom w shower room Glasgow Airport: 46 miles Glasgow city centre: 56 miles Directions From Glasgow follow the A82 passing Dumbarton and Balloch and follow signs for Loch Lomond. Turn left at Tarbet and on to the A83 and follow the road through Arrochar. At the top of the Rest and Be Thankful pass, turn left on the B828 towards Lochgoilhead. Follow the road along the edge of the loch for approximately 1 mile. Heathbank is on the left hand side. Situation Heathbank is situated in the sought after village of Lochgoilhead in Argyll. Lochgoilhead sits at the northern end of Loch Goil, where life centres around the village shop, restaurant and church. The area offers a spectacular landscape of mountains and lochs and is therefore an ideal base for outdoor enthusiasts. There is superb hill walking and climbing as well as excellent fishing and sailing on the loch. The drive to Lochgoilhead from Glasgow passing Loch Lomond, the Arrochar Alps and the Rest and Be Thankful, from where the single track road leads to Lochgoilhead, is a stunning journey. Description Heathbank is a detached, stone – built villa which occupies a prime elevated spot within the village, on the edge of Loch Goil. -

Upper Cormonachan Loch Goil, Argyll Archaeological Survey for July 2020
FIRAT ARCHAEOLOGICAL SERVICES Established 1992 Hillcroft, Station Road, Rhu, By Helensburgh, G84 8LW, Argyll, Scotland Upper Cormonachan Loch Goil, Argyll Archaeological Survey for July 2020 Director: Fiona M.C. Baker BA, FSAScot. Tel: 01436 820 334 Email: [email protected] Registered in Scotland. Company Number SC337432 Upper Cormonachan, Carrick Castle, Loch Goil, Argyll Contents Page Figure 1. Site location and plan 2 Figure 2. Plan of Cormonachan Woodlands path route. 3 Introduction 4 Planning Consent 4 Scope and Objectives of the Archaeological Work 5 Map Evidence 6 Figure 3. Timothy Pont map c. 1590 6 Figure 4. Roy Military Survey of Scotland 1747-55 7 Figure 5. First Edition Ordnance Survey map 1860 8 Figure 6. Second Edition Ordnance Survey map 1899 9 Figure 7. First and Second Edition Ordnance Survey maps 10 Documentary Evidence 11 Archaeological Background (Historic Environment Records) 16 Site Conditions and Impact of Development 23 Tree Felling 24 Figure 8: Site plan showing location of felled trees, path route and sign location 25 Site Description (includes condition assessment and mitigation) 26 Summary Conclusion 78 Recommendations 79 Interpretation board 79 Paths 80 Discovery and Excavation in Scotland entry 81 Photograph List 82 Drawing List 86 Bibliography and Map List 86 Reporting and Contents and Location of the Archive 87 Acknowledgements 87 Contact Addresses 88 Interpretation board figures Figure 9: Site plan showing path route and sign location 89 Figure 10: Site plan (no annotation) 90 Photographs with captions are present throughout the report. Firat Archaeological Services for Cormonachan Community Woodlands 1 Upper Cormonachan, Loch Goil, Argyll. -

Clyde Sea Lochs Trail
A great Clyde Sea Lochs Trail The Top Ten on the Clyde SeaLochs Trail day out! • Climb historic Dumbarton Castle Rock • Walk among the bluebells at Geilston Garden (early May) Birdwatch and walk at Ardmore Point The Clyde Sealochs Trail leads you on a fascinating • • Admire ‘The Hill House’ in Helensburgh 65 mile journey of discovery around the northern • Be inspired and nostalgic at Helensburgh Heroes shores of the Firth of Clyde and its beautiful sea Inspirational Centre and 1950’s Hucksters Café Clyde Sea lochs. • Explore Glenarn’s world class Rhododendron Gardens View Britain’s Largest Naval Base from the A817 ‘Haul Road’ Castles, gardens, history, wildlife, seashore and • • Time travel aboard PS Waverley, the world’s last sea going Lochs Trail woodland walks, coastal and mountain scenery, paddle steamer (Spring – Summer only) a Geocache trail, must see buildings and great • Discover Loch Long’s Viking heritage (Cove and Arrochar) places to eat – the Clyde Sea Lochs Trail has it all! • Climb ‘The Cobbler’ in the Arrochar Alps Use the map to see all the route possibilities and The Hill House information panel locations, most of which have nearby carparks and offer picnic spots with views. There is also a geocache trail directly linked to the route as an added attraction. Journeys to specific parts of the route can also be made by public transport. The trail is well signposted – just look out for the Clyde Sea Lochs Trail logo! Kilcreggan Pier Leaflet sponsored by: Balmillig B&B Friends of Loch Lomond and The Trossachs Visitor Giving Scheme Leaflet text and photos supplied by Loch Lomond Guides Front: Loch Long and the ‘Cobbler’ by Neil Wells Rhu MAP KEY Clyde Sea Lochs Trail map showing Seafront . -

Determination of the Off-Site Emergency Planning and Prior Information Areas for the Loch Goil Operational Berth
Determination of the Off-Site Emergency Planning and Prior Information Areas for the Loch Goil Operational Berth Radiation (Emergency Preparedness and Public Information) Regulations 2001 Project Assessment Report ONR-COP-PAR-16-024 Revision 0 8 February 2017 Office for Nuclear Regulation © Office for Nuclear Regulation, 2017 If you wish to reuse this information visit www.onr.org.uk/copyright for details. Published 02/17 For published documents, the electronic copy on the ONR website remains the most current publicly available version and copying or printing renders this document uncontrolled. Office for Nuclear Regulation Report ONR-COP-PAR-16-024 TRIM Ref: 2016/455694 EXECUTIVE SUMMARY Determination of the Off-site Emergency Planning and Prior Information Areas for the Loch Goil Operational Berth: Radiation (Emergency Preparedness and Public Information) Regulations 2001 The Office for Nuclear Regulation (ONR) is responsible for regulating the GB nuclear industry in order to protect the health and safety of employees and the public against risks of harm arising from ionising radiations. ONR is the regulatory authority for Radiation (Emergency Preparedness and Public Information) Regulations (REPPIR) 2001 on nuclear licensed sites, Ministry of Defence (MOD) Authorised nuclear sites and nuclear warship sites. ONR’s responsibilities include legal duties, where it is concluded that there is a potential for a reasonably foreseeable radiation emergency (as defined in REPPIR), to determine an off-site emergency planning area (i.e. the area within which, in ONR’s opinion, any member of the public is likely to be affected by such an emergency). In these cases, there is also a legal duty, under the same Regulations, for ONR to determine an area within which prior information is to be distributed to the public. -

Self Drive Boat Hire Loch Goil
20 1 2 3 4 CRAFT BEER Boat Trips THE ROYAL AN LOCHAN HOTEL TAP ROOM & SHOP 1 BREWERY TOURS TIGHNABRUAICH 2 Open 7 days, 10am-6pm 12 Sightseeing and fishing on the Kyles of Bute SIGHTSEEING AND FISHING ON THE KYLES OF BUTE 1hr £15 adult: £10 Kids Giles Ian Photo: PERIODS OF 1 HOUR £15 PER PERSON £10 FOR KIDS “A fantastic day, scenery is amazing. visitor CEntrE Contact Donald 07799661493 mausolEum & We saw Porpoises & Seals as well as lochsiDE Stunning lochsidehE BlooDymausoleum HighlanD and visitors’ Historyrgyll centre, with a rich catchingdcmarineboattrips.co.uk a life supply of Mackerel. bloody highlandt history and the afnal resting place of 9 theof InveraraythE DukEs Dukes of of Argyll. Excellent” ift shop Postcode PA21 2EA Accommodation, Restaurant & Lounge with outstanding sea views, Stunning viewsg and a warm welcomegarDEns – gift shop and refreshments. Contact Donald 07799661493 Spa Therapist, Barista Cofee, Shinty Bar, Free Moorings, 10 minutEs from BEnmorE ) s. Less than Openanbeautiful hour Thursday, from Benmore Inveraray Friday, Botanic SaturdayandApril Gardens.10 to Octoberminutes 10 - 4. from the Tel: 01499 600482 or Art Gallery, Non residents welcome www.dcmarineboattrips.co.uk Open 10-4 Thurs, Fri and Sat ( email [email protected] or phone 07887856514 to arrange other time Cairndow, Argyll, PA26 8BL PA21 2EA www.historickilmun.org www.fyneales.com t: 01700 811239 www.theroyalanlochan.co.uk charity SC SC041413 5 6 7 21 CrossroAds Award-winning food & Pet & GArden accommodation on the 7 shores of Loch Fyne LASERSPORT is played with authentic deactivated 12-bore shotguns. Guns fire an infrared beam at the clays and the electronic 19 scoreboard records the results SAFE ● FUN ● SUITABLE FOR ALL AGES & SKILL LEVELS ● ADDICTIVE Suitable for both Adults & Children Up to 5 people can shoot at the same time. -

@Issue013,May FINAL EDITION As PRINTED
www.hasteltd.co.uk THE WEE GOIL Loch Goil Community Newspaper £1 May 2012 Issue no. 13 £1 Sport Relief Loch Goil 2012 In our second year holding this event at Lochgoilhead, (with the closest other mile event being the flagship mile in Glasgow) we had some fifty entrants take on the challenge of running or walking 1, 3 or 6 miles. We had fantastic sunshine and some real personal achievements on the day, with our village legend John Egan (winner of the Steeple run 2011) coming home in first place closely followed by Jim Cairney who has recently started a new way of life with nutrition and exercise bringing a new sense of health and well being and his first medal award. Michelle Nobes completed her first event and 3 mile run and finished with a big smile and a medal worn with pride for the rest of the day. Continued on page 3 1 LETTER FROM THE EDITORS DATES for your DIARY Dear Readers, MAY We hope you like the revised front page to Saturday 5th bring you a bigger photograph. (And that Watersports Club Muster you will not miss the list of contents! - but also let us know if you do!) There are also Peace and Global Philosophy Study changes afoot in two of our local clubs, the Seminar Burns Club and the Women’s Rural, read all Castle of the Muses about them in this issue. Carrick Castle 1.00 - 5.00 pm A very big thank you to Roger Brook for his Sunday 6th four part series on the River Goil, the final Carrick Castle Daffodil Tea instalment is on page 11. -

On the Timing of Retreat of the Loch Lomond ('Younger Dryas
Lowe, J., Matthews, I., Mayfield, R., Lincoln, P., Palmer, A., Staff, R. and Timms, R. (2019) On the timing of retreat of the Loch Lomond (‘Younger Dryas’) Readvance icefield in the SW Scottish Highlands and its wider significance. Quaternary Science Reviews, 219, pp. 171-186. There may be differences between this version and the published version. You are advised to consult the publisher’s version if you wish to cite from it. http://eprints.gla.ac.uk/192781/ Deposited on: 12 August 2019 Enlighten – Research publications by members of the University of Glasgow http://eprints.gla.ac.uk On the timing of retreat of the Loch Lomond (‘Younger Dryas’) Readvance icefield in the SW Scottish Highlands and its wider significance John Lowe 1#, Ian Matthews 1, Roseanna Mayfield 1*, Paul Lincoln 1, Adrian Palmer1, Richard Staff 2 †, Rhys Timms 1 1 Centre for Quaternary Research (CQR), Department of Geography, Royal Holloway University of London, Egham, Surrey, UK, TW20 0EX. [email protected] ; [email protected] ; [email protected] 2 Oxford Radiocarbon Accelerator Unit (ORAU), University of Oxford, Dyson Perrins Building, South Parks Road, Oxford OX1 3QY *Current address: Department of Geography, Southampton University, University Road Southampton SO17 1BJ. [email protected] †Current address: Scottish Universities Environmental Research Centre (SUERC), University of Glasgow, Rankine Avenue, East Kilbride, UK, G75 0QF [email protected] # Corresponding author. [email protected] Abstract It has long been assumed that the last glacier expansion in the Scottish Highlands, the Loch Lomond Readvance (LLR), resulted from a cold reversal that was broadly coeval with the ‘Younger Dryas’ episode.