List Item Refludan : EPAR
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

201370Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 201370Orig1s000 MEDICAL REVIEW(S) M E M O R A N D U M DEPARTMENT OF HEALTH AND HUMAN SERVICES PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION CENTER FOR DRUG EVALUATION AND RESEARCH Date: July 8, 2011 From: Kathy M. Robie-Suh, M.D., Ph.D. Medical Team Leader Division of Hematology Products Subject: NDA 201370, resubmission April 11, 2011 Heparin Sodium Injection (heparin sodium, USP) Sponsor: Pfizer, Inc. 235 East 42nd Street New York, NY 10017-5755 To: NDA 201370 This is the second review cycle for this 505(b)(2) application for a new Heparin Sodium Injection product derived from porcine intestine. The presentations include several containing benzyl alcohol as a preservative and one preservative-free presentation. The first cycle review of the NDA found the application acceptable from a clinical viewpoint. However, manufacturing facilities inspection identified deficiencies that precluded approval and a Complete Response (CR) letter was issued on April 7, 2011. Labeling review was not completed at that time. In the current resubmission the sponsor has responded to the identified deficiencies deleting the two heparin supplier sites where deficiencies were found. The Amended Cross-Discipline Team Leader Review by Dr. Ali Al-Hakim (6/28/2011) comments that Office of Compliance has updated its recommendation in the Establishment Evaluation System and issued “ACCEPTABLE” overall recommendation for this NDA on June 27, 2011 and therefore the NDA is recommended for approval. No new clinical information is included in the resubmission. Clinical review of the original application and the resubmission was conducted by Dr. -

Pharmacoepidemiological.Study.Protocol.. ER1379468. A.Retrospective.Cohort.Study.To.Investigate.The.Initiation. And.Persistence.Of.Dual.Antiplatelet.Treatment.After
Pharmacoepidemiological.study.protocol.ER1379468. % . % % % % % Pharmacoepidemiological.study.protocol.. ER1379468. A.retrospective.cohort.study.to.investigate.the.initiation. and.persistence.of.dual.antiplatelet.treatment.after.. acute.coronary.syndrome.in.a.Finnish.setting.–.THALIA. % Author:(( ( ( Tuire%Prami( Protocol(number:(( %% ER1359468,%ME5CV51306( Sponsor:(( ( ( AstraZeneca%Nordic%Baltic% Protocol(version:(( ( 2.0( Protocol(date:(( ( ( 03%Jul%2014% ( . EPID%Research%Oy%. CONFIDENTIAL. % Pharmacoepidemiological.study.protocol.ER1379468... Version.2.0. 03.Jul.2014. Study Information Title% A% retrospective% cohort% study% to% investigate% the% initiation% and% persistence% of% dual% antiplatelet%treatment%after%acute%coronary%syndrome%in%a%Finnish%setting%–%THALIA% Protocol%version% ER1359468% identifier% ME5CV51306% EU%PAS%register% ENCEPP/SDPP/6161% number% Active%substance% ticagrelor%(ATC%B01AC24),%clopidogrel%(B01AC04),%prasugrel%(B01AC22)% Medicinal%product% Brilique,% Plavix,% Clopidogrel% accord,% Clopidogrel% actavis,% Clopidogrel% krka,% Clopidogrel% mylan,%Clopidogrel%orion,%Clopidogrel%teva%pharma,%Cloriocard,%Efient% Product%reference% N/A% Procedure%number% N/A% Marketing% AstraZeneca%Nordic%Baltic:%Brilique%(ticagrelor)% authorization% holder% financing%the%study% Joint%PASS% No% Research%question% To%describe%initiation%and%persistence%of%dual%antiplatelet%treatment%in%invasively%or%non5 and%objectives% invasively%treated%patients%hospitalized%for%acute%coronary%syndrome%% Country%of%study% Finland% Author% Tuire%Prami% -

Best Evidence for Antithrombotic Agent Reversal in Bleeding
3/16/2021 1 JAMES H. PAXTON, MD • DIRECTOR OF CLINICAL RESEARCH • DETROIT RECEIVING HOSPITAL (DRH), DEPARTMENT OF EMERGENCY MEDICINE, WAYNE STATE UNIVERSITY • ATTENDING PHYSICIAN • SINAI-GRACE HOSPITAL (SGH) • DETROIT RECEIVING HOSPITAL (DRH) 2 1 3/16/2021 DISCLOSURES • FUNDED RESEARCH SPONSORED BY: TELEFLEX INC, 410 MEDICAL, HOSPI CORP. 3 OBJECTIVES •AT THE CONCLUSION OF THIS LECTURE, THE LEARNER WILL: • RECOGNIZE COMMON ANTITHROMBOTIC MEDICATIONS ASSOCIATED WITH BLEEDING EMERGENCIES • BE ABLE TO EXPLAIN THE MECHANISMS OF ACTION FOR COMMON ANTITHROMBOTIC MEDICATIONS • UNDERSTAND THE EVIDENCE FOR RAPID REVERSAL OF ANTITHROMBOTIC MEDICATIONS, INCLUDING CONTROVERSIES AND BEST PRACTICES HAVE FEWER NIGHTMARES ABOUT SCENES LIKE THE ONE ON THE NEXT SLIDE? 4 2 3/16/2021 BLEEDING IS BAD 5 WHEN TO REVERSE? • WHEN RISK OF BLEEDING OUTWEIGHS RISK OF REVERSAL: • INTRACRANIAL HEMORRHAGE (ICH) • OTHER CNS HEMATOMA (E.G., INTRAOCULAR, SPINAL) • EXSANGUINATING GASTROINTESTINAL (GI) BLEED • UNCONTROLLED RETROPERITONEAL BLEED • HEMORRHAGE INTO EXTREMITY WITH COMPARTMENT SYNDROME • CONSIDER IN POSTERIOR EPISTAXIS, HEMOTHORAX, PERICARDIAL TAMPONADE 6 3 3/16/2021 ANTITHROMBOTIC AGENTS •ANTI-PLATELET DRUGS •ANTI-COAGULANTS •FIBRINOLYTICS 7 THROMBOGENESIS • Primary component in arterial thrombosis (“white clot”) • Target for antiplatelet drugs • Primary component in venous thrombosis (“red clot”) • Aggregated platelets + • Target for anticoagulants fibrin mesh • Target for fibrinolytics 8 4 3/16/2021 PLATELET AGGREGATION Antiplatelet Drugs Mechanism Aspirin* -

Melagatran Und Ecarinzeit.Pdf
Wednesday , August 06, 2003 1 Effects of Lepirudin, Argatroban and Melagatran and Additional Influence of Phenprocoumon on Ecarin Clotting Time Tivadar Fenyvesi* M.D., Ingrid Jörg* M.D., Christel Weiss + Ph.D., Job Harenberg* M.D. Key words: direct thrombin inhibitors, ecarin clotti ng time, oral anticoagulants, enhancing effects *IV. Department of Medicine +Institute for Biometrics and Medical Statistics University Hospital Mannheim Theodor Kutzer Ufer 1 - 3 68167 Mannheim Germany *Corresponding author Phone: +49 -621 -383 -3378 E-ma il: tivadar.fenyvesi @med1.ma.uni -heidelberg.de Version 06.08.03 for Thromb Res Review Copy Elsevier 1 of 22 Wednesday , August 06, 2003 2 Abstract Introduction: Direct thrombin inhibitors (DTI) prolong the ecarin clotting time (ECT). Oral anticoagulants (OA) decrease prothrombin levels and thus interacts with actions of DTIs on the ECT method during concomitant therapy. Materials and methods: Actions of lepirudin, argatroban, and melagatran on ECT were investigated in normal plasma (NP) and in plasma of patients (n = 23 each) on stable therapy with phenprocoumon (OACP). Individual line characteristics were tested statistically. Results: Control ECT in OACP was prolonged compared to NP (50.1±0.9 vs. 45.7±0.8 sec; p < 0.001). Lepirudin prolonged the ECT linearly. Argatroban and melagatran delivered biphasic dose-response curves. OA showed additive effects on the ECT of lepirudin but not of argatroban and melagatran. Both in NP and OACP the first and second slopes of melagatran were steeper compared to argatroban (primary analysis; p<0.001). When using the same drug, slopes in OACP were steeper than in NP (secondary analysis; p<0.001). -

Refludan, INN-Lepirudin
ANNEXE I autorisé RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT plus n'est médicament Ce 1 1. DENOMINATION DU MEDICAMENT Refludan, 20 mg, poudre pour solution injectable ou pour perfusion 2. COMPOSITION QUALITATIVE ET QUANTITATIVE Chaque flacon contient 20 mg de lépirudine. (La lépirudine est un produit ADN recombinant dérivé de cellules de levure) Pour une liste complète des excipients, voir rubrique 6.1. 3. FORME PHARMACEUTIQUE Poudre pour solution injectable ou pour perfusion. Poudre lyophilisée blanche à presque blanche. 4. DONNEES CLINIQUES 4.1 Indications thérapeutiques autorisé Inhibition de la coagulation chez des patients adultes atteints d'une thrombopénie induite par l'héparine (TIH) de type II et de maladie thrombo-embolique nécessitant un traitement antithrombotique par voie parentérale. plus Le diagnostic devrait être confirmé par le test d'activation plaquettaire induite par l'héparine (HIPAA = Heparin Induced Platelet Activation Assay) ou un test équivalent. n'est 4.2 Posologie et mode d’administration Le traitement par Refludan devrait être débuté sous le contrôle d'un médecin ayant une expérience des troubles de l’hémostase. Posologie initiale Inhibition de la coagulation chezmédicament des patients adultes atteints d'une TIH de type II et de maladie thrombo-embolique : Ce 0,4 mg/kg de poids corporel en bolus intraveineux, suivi de 0,15 mg/kg de poids corporel/heure en perfusion intraveineuse continue pendant 2 à 10 jours, voire plus si l'état clinique du patient le nécessite. La posologie dépend du poids corporel du patient jusqu'à 110 kg. Chez les patients pesant plus de 110 kg, la posologie ne doit pas dépasser celle calculée pour les patients pesant 110 kg (voir aussi tableaux 2 et 3 ci-après). -

Neuroradiology Fine Needle Spinal Procedures and Anticoagulation: Fine Needle Spinal Procedure: 20 Gauge Needle Or Smaller
Neuroradiology Fine Needle Spinal Procedures and Anticoagulation: Fine needle spinal procedure: 20 gauge needle or smaller. Typically 20 or 22 gauge is used. Medication Recommendation Aspirin & NSAIDS -Does NOT need to be stopped. Warfarin -Stop 5 days prior. -Check INR: INR ≤ 1.2 is normal. Proceed. *INR of 1.3 or 1.4 requires 24 hrs of post procedure monitoring. INR of ≥1.5LP not done. Thrombolytics (tPA) Unsafe. Should not be used pre or post procedure. Time frame uncertain. Heparin SQ (DVT prophylaxis) -Check platelets if on heparin ≥5 days. 5000 units SQ BID -No contraindication or delay if standard dosing of 5000 units SQ BID. -If given ≥ TID or > 5,000 units/dose or > 10,000 units /day: follow heparin IV protocol. Heparin IV -Wait 2-4 hours after last dose of IV heparin. -Document normal PTT. -Check platelets if on heparin ≥5 days. -Wait at least 1 hr after procedure to restart heparin drip. LMWH (Enoxaparin) prophylaxis -Wait 12 hrs after last dose to perform procedure. 30 mg SQ BID or -Wait 24 hrs after LP to restart med. 40 mg SQ daily LMHW (Enoxaparin) treatment -Wait 24 hrs after last dose to perform procedure. 1-1.5 mg/kg/day -Wait 24 hrs to restart med. Fondaparinux (Arixtra) -Withhold based on Cr. Cr Cl >50 wait 2-3 days; Cr Cl < 50 wait 3-5 days Clopidogrel (Plavix) Stop 5 days prior. Ticlopidine (Ticlid) Stop 14 days prior. Abciximab (ReoPro) (IIb/IIIa) Stop 2 days prior. Eptifibatide (Integrilin) (IIb/IIIa) Stop 4-8 HOURS prior. Tirofiban (Aggrastat) (IIb/IIIa) Stop 8 HOURS prior. -

Cleveland Clinic Anticoagulation Management Program (C-Camp)
CLEVELAND CLINIC ANTICOAGULATION MANAGEMENT PROGRAM (C-CAMP) 1 Table of Contents I. EXECUTIVE SUMMARY ......................................................................................................................................... 6 II. VENOUS THROMBOEMBOLISM RISK ASSESSMENT AND PROPHYLAXIS ............................................. 9 III. RECOMMENDED PROPHYLAXIS OPTIONS FOR THE PREVENTION OF VENOUS THROMBOEMBOLISM. ........................................................................................................................................ 10 IIIA. RECOMMENDED PROPHYLAXIS OPTIONS FOR THE PREVENTION OF VENOUS THROMBOEMBOLISM BASED ON RISK FACTOR ASSESSMENT ............................................................. 12 A) UNFRACTIONATED HEPARIN (UFH) ................................................................................................................... 14 B) LOW MOLECULAR WEIGHT HEPARIN (LMWH) ENOXAPARIN (LOVENOX®) .............................................. 14 C) FONDAPARINUX/ (ARIXTRA®) …………………………………………………………………………..……16 D) RIVAROXABAN (XARELTO®) .......................................................................................................................... 16 E) DESIRUDIN (IPRIVASK®)………………………………………………………………………………..…….17 F) WARFARIN/COUMADIN®) ............................................................................................................................... 16 G) ASPIRIN……………………………………………………………………………………………………..……18 H) INTERMITTENT PNEUMATIC COMPRESSION DEVICES ..................................................................................... -
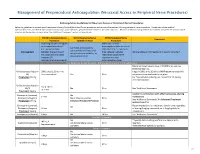
Neuraxial Or Nerve Procedures
Management of Periprocedural Anticoagulation (Neuraxial Access or Peripheral Nerve Procedures) Anticoagulation Guidelines for Neuraxial Access or Peripheral Nerve Procedures Below are guidelines to prevent spinal hematoma following Epidural/Intrathecal/Spinal procedures and perineural hematoma following peripheral nerve procedures. Procedures include epidural injections/infusions, intrathecal injections/infusions/pumps, spinal injections, peripheral nerve catheters, and plexus infusions. Decisions to deviate from guideline recommendations given the specific clinical situation are the decision of the provider. See ‘Additional Comments’ section for more details. PRIOR to Neuraxial/Nerve WHILE Neuraxial/Nerve AFTER Neuraxial/Nerve Comments Procedure Catheter in Place Procedure How long should I hold prior When can I restart to neuraxial procedure? anticoagulants after neuraxial Can I give anticoagulants (i.e., minimum time procedures? (i.e., minimum concurrently with neuraxial, Anticoagulant between the last dose of time between catheter What additional information do I need to consider? peripheral nerve catheter, or anticoagulant and spinal removal or spinal/nerve plexus placement? injection OR injection and next neuraxial/nerve placement) anticoagulation dose) Low-Molecular Weight Heparins, Unfractionated Heparin, and Fondaparinux Maximum total heparin dose of 10,000 units per day (5000 SQ Q12 hrs) Unfractionated Heparin 5000 units Q 12 hrs – no Heparin 5000 units SQ 8 hrs is NOT recommended with SQ time restrictions Yes 2 hrs concurrent -

Collaborative Study on Monitoring Methods to Determine Direct Thrombin Inhibitors Lepirudin and Argatroban
Collaborative Study On Monitoring Methods To Determine Direct Thrombin Inhibitors Lepirudin And Argatroban On behalf of the Control of Anticoagulation Subcommittee of the Scientific and Standardization Committee of the International Society of Thrombosis and Haemostasis E. Gray 1 and J Harenberg * SUMMARY Background: Direct thrombin inhibitors are now used clinically for the prophylaxis and treatment of thrombosis and related cardiovascular diseases. Although all these inhibitors bind to thrombin, their modes of action are different. Routine anticoagulant monitoring tests such as the APTT are used to estimate the activity of these inhibitors. However, no studies have been carried out to assess the suitability of these tests. Methods: An international collaborative study was carried out utilising a panel of plasmas spiked with lepirudin and argatroban to evaluate the robustness and the sensitivity of the different monitoring methods. Results: Thirteen laboratories took part in the trial. The point-of-care TAS-analyser, using cards with high or low concentrations of ecarin reagents gave the lowest intra- and inter- laboratory variation. APTT using local or common APTT reagents also showed acceptable within and between-laboratory variations. The chromogenic anti-IIa activity and the wet ecarin clotting time gave higher inter-laboratory variations. The ELISA for lepirudin had the highest inter-laboratory variation. Conclusion: APTT and the TAS-analyser with ECT-cards gave the most reproducible results compared to other methods. Further studies will evaluate the validity for patient samples. INTRODUCTION Direct and indirect serine protease inhibitors are used clinically for inhibition of blood coagulation (1). Direct inhibitors such as hirudin and fondaparinux inhibit individual serine proteases such as thrombin and factor Xa. -

RBCH VTE Prevention Guidelines
VTE Prevention Guidelines (Venous Thromboembolism) (Venous Thromboembolism) When using this document please ensure that the version you are using is the most up to date either by checking on the Trust intranet or if the review date has passed, please contact the author. ‘Out of date policy documents must not be relied upon’ Approval Version Issue Date Review Date Document Author Committee Drugs & 1.2 July 2010 July 2012 Dr Joseph Chacko Therapeutics Consultant Haematologist Jacqui Bowden Clinical Pharmacy Manager D&TC 2 July 2012 July 2014 Kareena Marotta & Jason Mainwaring Version Control Version Date Author Section Principle Amendment Changes 2 July 2012 Kareena Changed Enoxaparin to Dalteparin Marotta & Amended HIT monitoring guidance Jason Changed Lepirudin to Danaparoid Mainwaring Added audit section VTE Prevention Guidelines – Version 2 Page 1 of 22 July 2012 Contents 3 Introduction 3 Definitions and abbreviations 4 Assessing risks of VTE and bleeding 5 Care pathway 6 Using VTE prophylaxis Pharmacological Mechanical 9 Medical patients 10 Stroke 11 Cancer and patients with a central venous catheter 12 Palliative care 13 Non-orthopaedic surgery 14 Orthopaedic surgery 15 Critical care 16 Pregnancy and post-partum – see separate guidelines: Reducing the risk and management of venous thromboembolism (VTE) in pregnancy 17 Planning for discharge 18 Monitoring, diagnosing and managing heparin-induced thrombocytopaenia (HIT) 21 References 22 Appendices I. Risk assessment for venous thromboembolism (VTE) for adult patients admitted to hospital (page 2 of the Acute Prescription and Administration Record) II. Guide to using the VTE Risk Assessment Form III. Preventing blood clots in hospital (patient information leaflet) IV. -

Protocol for Non-Interventional Studies Based on Existing Data TITLE PAGE Document Number: C30445781-01
ABCD Protocol for non-interventional studies based on existing data TITLE PAGE Document Number: c30445781-01 BI Study Number: 1237-0090 BI Investigational Stiolto® Respimat® Product(s): The Role of Inhaler Device in the Treatment Persistence with Dual Title: Bronchodilators in Patients with COPD Protocol version 1.0 identifier: Date of last version of N/A protocol: PASS: No EU PAS register Study not registered number: Olodaterol and Tiotropium Bromide (ATC R03AL06) Active substance: Umeclidinium and Vilanterol (ATC R03AL03) Medicinal product: Stiolto® Respimat®; Anoro® Ellipta® Product reference: N/A Procedure number: N/A Joint PASS: No The primary objective of the study is to use US data to determine relative persistence between Olodaterol/Tiotropium Bromide delivered with the Respimat soft mist inhaler and Umeclidinium/Vilanterol delivered with the Ellipta dry powder inhaler using a 1:2 propensity score matched analysis. The secondary objectives of the study are as follows: Research question and - Characterize new users of Olodaterol/Tiotropium Bromide objectives: and Umeclidinium/Vilanterol in terms of demographics, medication use, comorbidities, and other variables, before and after propensity score matching. - Determine the incidence rate and proportion of patients discontinuing or switching among new users of Olodaterol/Tiotropium Bromide and Umeclidinium/ Vilanterol. - Determine the relative rate and proportion of patients discontinuing between Olodaterol/ Tiotropium Bromide and 001-MCG-102_RD-02 (1.0) / Saved on: 22 Oct 2015 Boehringer Ingelheim Page 2 of 69 Protocol for non-interventional studies based on existing data BI Study Number 1237-0090 c30445781-01 Proprietary confidential information © 2019 Boehringer Ingelheim International GmbH or one or more of its affiliated companies Umeclidinium/Vilanterol. -

Dear Health Care Professional Letter Refludan 2012 Final
May 31, 2012 Discontinuation of Refludan ® [lepirudin (rDNA) for injection] Dear Health Care Professional: After careful consideration, Bayer HealthCare would like to inform you that we will discontinue marketing Refludan ® [lepirudin (rDNA) for injection] on May 31, 2012 and no further product is expected to be shipped to wholesalers in the United States after that date. It is important to emphasize that these actions are not related to safety concerns. Bayer HealthCare has had a manufacturing and supply agreement in place with a third-party manufacturer that permitted the sale and distribution of Refludan ® by Bayer in the U.S. The third- party manufacturer of Refludan ® has permanently ceased production of the product, which has led to the product’s discontinuation in the U.S. Bayer has notified the Food and Drug Administration that no further product is expected to be shipped to wholesalers in the U.S. as of May 31, 2012. Bayer anticipates the supply of Refludan ® will be depleted by mid-2013. Refludan ® [lepirudin (rDNA) for injection] is indicated for anticoagulation in patients with heparin- induced thrombocytopenia (HIT) and associated thromboembolic disease in order to prevent further thromboembolic complications (TEC). 1 We encourage you to discuss treatment alternatives with your patients. Recommendations for use of anticoagulants other than Refludan ® are described in treatment guidelines for HIT. 2 We regret any inconvenience this decision may have on your practice and we very much appreciate your support for Refludan ®. Should you have any questions, please call Bayer Customer Service (877-229-3750) or Bayer Medical Communications (888-842-2937).