Beneficial Effects of Caffeine Consumption on Diabetes-Induced Alterations in the Hippocampus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanisms of Synaptic Plasticity Mediated by Clathrin Adaptor-Protein Complexes 1 and 2 in Mice
Mechanisms of synaptic plasticity mediated by Clathrin Adaptor-protein complexes 1 and 2 in mice Dissertation for the award of the degree “Doctor rerum naturalium” at the Georg-August-University Göttingen within the doctoral program “Molecular Biology of Cells” of the Georg-August University School of Science (GAUSS) Submitted by Ratnakar Mishra Born in Birpur, Bihar, India Göttingen, Germany 2019 1 Members of the Thesis Committee Prof. Dr. Peter Schu Institute for Cellular Biochemistry, (Supervisor and first referee) University Medical Center Göttingen, Germany Dr. Hans Dieter Schmitt Neurobiology, Max Planck Institute (Second referee) for Biophysical Chemistry, Göttingen, Germany Prof. Dr. med. Thomas A. Bayer Division of Molecular Psychiatry, University Medical Center, Göttingen, Germany Additional Members of the Examination Board Prof. Dr. Silvio O. Rizzoli Department of Neuro-and Sensory Physiology, University Medical Center Göttingen, Germany Dr. Roland Dosch Institute of Developmental Biochemistry, University Medical Center Göttingen, Germany Prof. Dr. med. Martin Oppermann Institute of Cellular and Molecular Immunology, University Medical Center, Göttingen, Germany Date of oral examination: 14th may 2019 2 Table of Contents List of abbreviations ................................................................................. 5 Abstract ................................................................................................... 7 Chapter 1: Introduction ............................................................................ -

Adenosine Enhances Sweet Taste Through A2B Receptors in the Taste Bud
322 • The Journal of Neuroscience, January 4, 2012 • 32(1):322–330 Cellular/Molecular Adenosine Enhances Sweet Taste through A2B Receptors in the Taste Bud Robin Dando,1* Gennady Dvoryanchikov,1* Elizabeth Pereira,1 Nirupa Chaudhari,1,2 and Stephen D. Roper1,2 1Department of Physiology and Biophysics and 2Program in Neuroscience, Miller School of Medicine, University of Miami, Miami, Florida 33136 Mammalian taste buds use ATP as a neurotransmitter. Taste Receptor (type II) cells secrete ATP via gap junction hemichannels into the narrowextracellularspaceswithinatastebud.ThisATPexcitesprimarysensoryafferentfibersandalsostimulatesneighboringtastebud cells.HereweshowthatextracellularATPisenzymaticallydegradedtoadenosinewithinmousevallatetastebudsandthatthisnucleoside acts as an autocrine neuromodulator to selectively enhance sweet taste. In Receptor cells in a lingual slice preparation, Ca 2ϩ mobilization evoked by focally applied artificial sweeteners was significantly enhanced by adenosine (50 M). Adenosine had no effect on bitter or umami taste responses, and the nucleoside did not affect Presynaptic (type III) taste cells. We also used biosensor cells to measure transmitter release from isolated taste buds. Adenosine (5 M) enhanced ATP release evoked by sweet but not bitter taste stimuli. Using single-cellreversetranscriptase(RT)-PCRonisolatedvallatetastecells,weshowthatmanyReceptorcellsexpresstheadenosinereceptor, Adora2b, while Presynaptic (type III) and Glial-like (type I) cells seldom do. Furthermore, Adora2b receptors are significantly associated with expression of the sweet taste receptor subunit, Tas1r2. Adenosine is generated during taste stimulation mainly by the action of the ecto-5Ј-nucleotidase, NT5E, and to a lesser extent, prostatic acid phosphatase. Both these ecto-nucleotidases are expressed by Presyn- aptic cells, as shown by single-cell RT-PCR, enzyme histochemistry, and immunofluorescence. Our findings suggest that ATP released during taste reception is degraded to adenosine to exert positive modulation particularly on sweet taste. -

Adolescent Exposure to Δ9-Tetrahydrocannabinol Alters The
Molecular Psychiatry https://doi.org/10.1038/s41380-018-0243-x ARTICLE Adolescent exposure to Δ9-tetrahydrocannabinol alters the transcriptional trajectory and dendritic architecture of prefrontal pyramidal neurons 1,2,3,4,6 1,2,3,4 1,4 1,4 Michael L. Miller ● Benjamin Chadwick ● Dara L. Dickstein ● Immanuel Purushothaman ● 1,2,3,4 1,2,3,4 1,2,4 1,4 2,4,5 Gabor Egervari ● Tanni Rahman ● Chloe Tessereau ● Patrick R. Hof ● Panos Roussos ● 1,4 1,4 1,2,4 Li Shen ● Mark G. Baxter ● Yasmin L. Hurd Received: 5 February 2017 / Revised: 25 July 2018 / Accepted: 8 August 2018 © The Author(s) 2018. This article is published with open access Abstract Neuronal circuits within the prefrontal cortex (PFC) mediate higher cognitive functions and emotional regulation that are disrupted in psychiatric disorders. The PFC undergoes significant maturation during adolescence, a period when cannabis use in humans has been linked to subsequent vulnerability to psychiatric disorders such as addiction and schizophrenia. Here, we investigated in a rat model the effects of adolescent exposure to Δ9-tetrahydrocannabinol (THC), a psychoactive 1234567890();,: 1234567890();,: component of cannabis, on the morphological architecture and transcriptional profile of layer III pyramidal neurons—using cell type- and layer-specific high-resolution microscopy, laser capture microdissection and next-generation RNA- sequencing. The results confirmed known normal expansions in basal dendritic arborization and dendritic spine pruning during the transition from late adolescence to early adulthood that were accompanied by differential expression of gene networks associated with neurodevelopment in control animals. In contrast, THC exposure disrupted the normal developmental process by inducing premature pruning of dendritic spines and allostatic atrophy of dendritic arborization in early adulthood. -

I REGENERATIVE MEDICINE APPROACHES to SPINAL CORD
REGENERATIVE MEDICINE APPROACHES TO SPINAL CORD INJURY A Dissertation Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Ashley Elizabeth Mohrman March 2017 i ABSTRACT Hundreds of thousands of people suffer from spinal cord injuries in the U.S.A. alone, with very few patients ever experiencing complete recovery. Complexity of the tissue and inflammatory response contribute to this lack of recovery, as the proper function of the central nervous system relies on its highly specific structural and spatial organization. The overall goal of this dissertation project is to study the central nervous system in the healthy and injured state so as to devise appropriate strategies to recover tissue homeostasis, and ultimately function, from an injured state. A specific spinal cord injury model, syringomyelia, was studied; this condition presents as a fluid filled cyst within the spinal cord. Molecular evaluation at three and six weeks post-injury revealed a large inflammatory response including leukocyte invasion, losses in neuronal transmission and signaling, and upregulation in important osmoregulators. These included osmotic stress regulating metabolites betaine and taurine, as well as the betaine/GABA transporter (BGT-1), potassium chloride transporter (KCC4), and water transporter aquaporin 1 (AQP1). To study cellular behavior in native tissue, adult neural stem cells from the subventricular niche were differentiated in vitro. These cells were tested under various culture conditions for cell phenotype preferences. A mostly pure (>80%) population of neural stem cells could be specified using soft, hydrogel substrates with a laminin coating and interferon-γ supplementation. -

The Synapse As a Central Target for Neurodevelopmental Susceptibility to Pesticides
toxics Review The Synapse as a Central Target for Neurodevelopmental Susceptibility to Pesticides Aimee Vester 1 and W. Michael Caudle 1,2,* 1 Department of Environmental Health, Rollins School of Public Health, Emory University, Atlanta, GA 30322, USA; [email protected] 2 Center for Neurodegenerative Disease, School of Medicine, Emory University, Atlanta, GA 30322, USA * Correspondence: [email protected]; Tel.: +1-404-712-8432 Academic Editor: David R. Wallace Received: 30 June 2016; Accepted: 17 August 2016; Published: 26 August 2016 Abstract: The developmental period of the nervous system is carefully orchestrated and highly vulnerable to alterations. One crucial factor of a properly-functioning nervous system is the synapse, as synaptic signaling is critical for the formation and maturation of neural circuits. Studies show that genetic and environmental impacts can affect diverse components of synaptic function. Importantly, synaptic dysfunction is known to be associated with neurologic and psychiatric disorders, as well as more subtle cognitive, psychomotor, and sensory defects. Given the importance of the synapse in numerous domains, we wanted to delineate the effects of pesticide exposure on synaptic function. In this review, we summarize current epidemiologic and molecular studies that demonstrate organochlorine, organophosphate, and pyrethroid pesticide exposures target the developing synapse. We postulate that the synapse plays a central role in synaptic vulnerability to pesticide exposure during neurodevelopment, and the synapse is a worthy candidate for investigating more subtle effects of chronic pesticide exposure in future studies. Keywords: dopamine; GABA; glutamate; neurodevelopment; neurotoxicity; organochlorine; organophosphate; pesticide; pyrethroid; synapse 1. Neurodevelopment Neurodevelopment is comprised of a highly orchestrated sequence of biological processes, including proliferation, migration, differentiation, and synaptogenesis, which must occur for the normal development and function of the nervous system. -
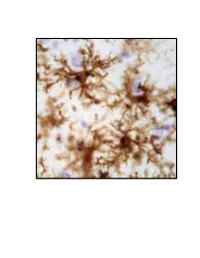
Neuroinflammation, Glutamate Regulation and Memory
Neuroinflammation, Glutamate Regulation and Memory. DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Holly M. Brothers, M.A. Graduate Program in Psychology The Ohio State University 2013 Dissertation Committee: Gary G. Berntson, John P. Bruno, Phillip G. Popovich, and Gary L. Wenk Copyrighted by Holly Marie Brothers 2013 Abstract Neuroinflammation and excessive glutamatergic signaling have deleterious effects in the brain, are mutually promoting, and play a role in the onset and progression of neurodegenerative diseases. It was my goal to better understand the relationship between neuroinflammation, glutamate dysregulation and clinical symptoms, as well as identify potential therapeutic targets. To do this, I studied molecular, cellular and behavioral outcomes across various time points in young rats with experimentally-induced chronic neuroinflammation and older rats with age-associated neuroinflammation. I focused on Alzheimer’s (AD) and Parkinson’s (PD), and tested only treatments that had therapeutic potential in a clinical setting. These studies led to a number of important discoveries, summarized below. I first investigated the effects of reducing pre-synaptic glutamate release by caffeine treatment, and discovered that caffeine attenuated experimentally-induced but not age- associated neuroinflammation and was not sufficient to improve cognitive performance (Chapter 2). During these investigations, I discovered that young rats with experimentally- induced neuroinflammation reared on their hind legs less, a behavioral change associated with PD. Therefore, I next expanded the scope of my studies to include a thorough investigation of brain systems that degenerate in PD, the basal ganglia and brainstem, using our model of chronic neuroinflammation (Chapter 3). -

Differential Regulation of Synaptic AP-2/Clathrin Vesicle Uncoating In
www.nature.com/scientificreports OPEN Differential regulation of synaptic AP-2/clathrin vesicle uncoating in synaptic plasticity Received: 9 June 2017 Ermes Candiello1, Ratnakar Mishra1, Bernhard Schmidt1, Olaf Jahn2 & Peter Schu1 Accepted: 24 October 2017 AP-1/ 1B-deficiency causes X-linked intellectual disability. AP-1/ 1B / mice have impaired synaptic Published: xx xx xxxx σ σ − − vesicle recycling, fewer synaptic vesicles and enhanced endosome maturation mediated by AP-1/σ1A. Despite defects in synaptic vesicle recycling synapses contain two times more endocytic AP-2 clathrin- coated vesicles. We demonstrate increased formation of two classes of AP-2/clathrin coated vesicles. One which uncoats readily and a second with a stabilised clathrin coat. Coat stabilisation is mediated by three molecular mechanisms: reduced recruitment of Hsc70 and synaptojanin1 and enhanced μ2/ AP-2 phosphorylation and activation. Stabilised AP-2 vesicles are enriched in the structural active zone proteins Git1 and stonin2 and synapses contain more Git1. Endocytosis of the synaptic vesicle exocytosis regulating Munc13 isoforms are differentially effected. Regulation of synaptic protein endocytosis by the differential stability of AP-2/clathrin coats is a novel molecular mechanism of synaptic plasticity. AP-1 and AP-2 clathrin adaptor-protein complexes have essential functions in synaptic vesicle (SV) recycling. AP-2 clathrin-mediated-endocytosis (CME) facilitates SV endocytosis, AP-1 complexes mediate TGN/endosome protein sorting via clathrin-coated-vesicles (CCV). In brain two γ1AP-1 complexes exist, which share β1 and μ1A, but differ in their σ1 adaptins. The ubiquitous AP-1 contains σ1A, the tissue-specific AP-1 contains σ1B. Deficiency of the X-chromosome encoded σ1B causes severe mental retardation1. -

Caffeine Consumption Attenuates Neurochemical Modifications in The
JOURNAL OF NEUROCHEMISTRY | 2009 | 111 | 368–379 doi: 10.1111/j.1471-4159.2009.06349.x , , , , *Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal Laboratory for functional and metabolic imaging (LIFMET), Center for Biomedical Imaging (CIBM), Ecole Polytechnique Fe´de´rale de Lausanne (EPFL), Lausanne, Switzerland àDepartment of Biochemistry, Faculty of Sciences and Technology, University of Coimbra, Coimbra, Portugal §Institute of Biochemistry, Faculty of Medicine, University of Coimbra, Coimbra, Portugal ¶Departments of Radiology, University of Geneva and University of Lausanne, Lausanne, Switzerland Abstract p < 0.05) and taurine (23 ± 4%, p < 0.01), supporting the Type 1 diabetes can affect hippocampal function triggering ability of caffeine to control osmoregulation. Compared to cognitive impairment through unknown mechanisms. Caf- controls, the hippocampus of diabetic rats displayed a re- feine consumption prevents hippocampal degeneration and duced density of synaptic proteins syntaxin, synaptophysin memory dysfunction upon different insults and is also known and synaptosome-associated protein of 25 kDa (in average to affect peripheral glucose metabolism. Thus we now 18 ± 1%, p < 0.05) as well increased glial fibrillary acidic characterized glucose transport and the neurochemical protein (20 ± 5%, p < 0.05), suggesting synaptic degenera- profile in the hippocampus of streptozotocin-induced dia- tion and astrogliosis, which were prevented by caffeine betic rats using in vivo 1H NMR spectroscopy and tested consumption. In conclusion, neurochemical alterations in the the effect of caffeine consumption thereupon. We found that hippocampus of diabetic rats are not related to defects of hippocampal glucose content and transport were unaltered glucose transport but likely reflect osmoregulatory adapta- in diabetic rats, irrespective of caffeine consumption. -

SNAP-25 Is a Component of a Ubiquitous SNARE Complex Required for Evoked Neuroexocytosis in Gabaergic Neurons Lawrence C
University of New Mexico UNM Digital Repository Biomedical Sciences ETDs Electronic Theses and Dissertations 5-1-2011 SNAP-25 is a component of a ubiquitous SNARE complex required for evoked neuroexocytosis in GABAergic neurons Lawrence C. R. Tafoya Follow this and additional works at: https://digitalrepository.unm.edu/biom_etds Recommended Citation Tafoya, Lawrence C. R.. "SNAP-25 is a component of a ubiquitous SNARE complex required for evoked neuroexocytosis in GABAergic neurons." (2011). https://digitalrepository.unm.edu/biom_etds/33 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biomedical Sciences ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. i ii Dedication I dedicate this dissertation to my beautiful wife Charlene and my two precious daughters, Isabella and Gabriella. It is through their unconditional support, undying patience, and infinite understanding that I was able to reach this important milestone. I cannot begin to repay them for the numerous sacrifices they gracefully made over the years. Individually, I would like to thank my oldest daughter, Isabella, for her inquisitive nature, serving as a continual reminder that knowledge belongs to those who seek it and, at any age, one should never stop questioning the world around us. For Gracie, I owe my gratitude for the moments of levity she provides, which extinguish panic, neutralize anxiety, and put into perspective the true priorities in life. Lastly, I pay tribute to my wife, who single-handedly has galvanized my success as a father, husband, physician, researcher, and a man. -

Delineating the Efficacy of a Cannabis-Based Medicine at Advanced
Journal of Alzheimer’s Disease 54 (2016) 903–912 903 DOI 10.3233/JAD-160533 IOS Press Delineating the Efficacy of a Cannabis-Based Medicine at Advanced Stages of Dementia in a Murine Model Ester Asoa,b,∗, Pol Andres-Benito´ a,b and Isidro Ferrera,b aInstitut de Neuropatologia, Servei d’Anatomia Patol`ogica, IDIBELL-Hospital Universitari de Bellvitge, Universitat de Barcelona, L’Hospitalet de Llobregat, Spain bCIBERNED, Centro de Investigaci´on Biom´edica en Red de Enfermedades Neurodegenerativas, Instituto Carlos III, Spain Handling Associate Editor: Tommaso Cassano Accepted 19 June 2016 Abstract. Previous reports have demonstrated that the combination of 9-tetrahydrocannabinol (9-THC) and cannabidiol (CBD) botanical extracts, which are the components of an already approved cannabis-based medicine, reduce the Alzheimer- like phenotype of APP/PS1 transgenic mice when chronically administered during the early symptomatic stage. Here, we provide evidence that such natural cannabinoids are still effective in reducing memory impairment in APP/PS1 mice at advanced stages of the disease but are not effective in modifying the A processing or in reducing the glial reactivity associated with aberrant A deposition as occurs when administered at early stages of the disease. The present study also demonstrates that natural cannabinoids do not affect cognitive impairment associated with healthy aging in wild-type mice. The positive effects induced by 9-THC and CBD in aged APP/PS1 mice are associated with reduced GluR2/3 and increased levels of GABA-A -

Caffeine Acts Through Neuronal Adenosine A2A Receptors to Prevent Mood and Memory Dysfunction Triggered by Chronic Stress
Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress Manuella P. Kastera,b,1, Nuno J. Machadoa,1, Henrique B. Silvaa,1, Ana Nunesa,1, Ana Paula Ardaisa,c, Magda Santanaa, Younis Baqid,e, Christa E. Müllerd, Ana Lúcia S. Rodriguesb, Lisiane O. Porciúnculac, Jiang Fan Chenf, Ângelo R. Toméa,g, Paula Agostinhoa,h, Paula M. Canasa, and Rodrigo A. Cunhaa,h,2 aCNC-Center for Neuroscience and Cell Biology, University of Coimbra, 3004-504 Coimbra, Portugal; bDepartment of Biochemistry, Federal University of Santa Catarina, Florianópolis, Santa Catarina, Brazil; cDepartament of Biochemistry, Federal University of Rio Grande do Sul, 90035-003 Porto Alegre, Rio Grande do Sul, Brazil, dPharmaceutical Institute, University of Bonn, D-53121 Bonn, Germany; eDepartment of Chemistry, Sultan Qaboos University, 123 Muscat, Oman; fDepartment of Neurology and Pharmacology, Boston University School of Medicine, Boston, MA 02118; gDepartment of Life Sciences, University of Coimbra, 3004-517 Coimbra, Portugal; and hFaculty of Medicine, University of Coimbra, 3004-504 Coimbra, Portugal Edited by Bruce S. McEwen, The Rockefeller University, New York, NY, and approved May 11, 2015 (received for review December 3, 2014) The consumption of caffeine (an adenosine receptor antagonist) to control aberrant plasticity (11, 12) and synaptotoxicity (13–15) or correlates inversely with depression and memory deterioration, from A2AR’s impact on astrocytes (16) or microglia (17). The and adenosine A2A receptor (A2AR) antagonists emerge as candi- protection provided by A2AR blockade prompts the hypothesis that date therapeutic targets because they control aberrant synaptic A2AR antagonism may underlie the beneficial effects of caffeine plasticity and afford neuroprotection. -

Review Article the Histone Modifications of Neuronal Plasticity
Hindawi Neural Plasticity Volume 2021, Article ID 6690523, 7 pages https://doi.org/10.1155/2021/6690523 Review Article The Histone Modifications of Neuronal Plasticity Huixia Geng ,1 Hongyang Chen ,2 Haiying Wang ,2 and Lai Wang 1,2 1Institute of Chronic Disease Risks Assessment, School of Nursing and Health Sciences, Henan University, Kaifeng, 475004 Henan Province, China 2College of Life Science, Henan University, Kaifeng, 475004 Henan Province, China Correspondence should be addressed to Lai Wang; [email protected] Received 17 December 2020; Revised 21 January 2021; Accepted 30 January 2021; Published 11 February 2021 Academic Editor: Wei-Lin Liu Copyright © 2021 Huixia Geng et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Nucleosomes composed of histone octamer and DNA are the basic structural unit in the eukaryote chromosome. Under the stimulation of various factors, histones will undergo posttranslational modifications such as methylation, phosphorylation, acetylation, and ubiquitination, which change the three-dimensional structure of chromosomes and affect gene expression. Therefore, the combination of different states of histone modifications modulates gene expression is called histone code. The formation of learning and memory is one of the most important mechanisms for animals to adapt to environmental changes. A large number of studies have shown that histone codes are involved in the formation and consolidation of learning and memory. Here, we review the most recent literature of histone modification in regulating neurogenesis, dendritic spine dynamic, synapse formation, and synaptic plasticity.