Viewed in [41])
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeted Genes and Methodology Details for Neuromuscular Genetic Panels
Targeted Genes and Methodology Details for Neuromuscular Genetic Panels Reference transcripts based on build GRCh37 (hg19) interrogated by Neuromuscular Genetic Panels Next-generation sequencing (NGS) and/or Sanger sequencing is performed Motor Neuron Disease Panel to test for the presence of a mutation in these genes. Gene GenBank Accession Number Regions of homology, high GC-rich content, and repetitive sequences may ALS2 NM_020919 not provide accurate sequence. Therefore, all reported alterations detected ANG NM_001145 by NGS are confirmed by an independent reference method based on laboratory developed criteria. However, this does not rule out the possibility CHMP2B NM_014043 of a false-negative result in these regions. ERBB4 NM_005235 Sanger sequencing is used to confirm alterations detected by NGS when FIG4 NM_014845 appropriate.(Unpublished Mayo method) FUS NM_004960 HNRNPA1 NM_031157 OPTN NM_021980 PFN1 NM_005022 SETX NM_015046 SIGMAR1 NM_005866 SOD1 NM_000454 SQSTM1 NM_003900 TARDBP NM_007375 UBQLN2 NM_013444 VAPB NM_004738 VCP NM_007126 ©2018 Mayo Foundation for Medical Education and Research Page 1 of 14 MC4091-83rev1018 Muscular Dystrophy Panel Muscular Dystrophy Panel Gene GenBank Accession Number Gene GenBank Accession Number ACTA1 NM_001100 LMNA NM_170707 ANO5 NM_213599 LPIN1 NM_145693 B3GALNT2 NM_152490 MATR3 NM_199189 B4GAT1 NM_006876 MYH2 NM_017534 BAG3 NM_004281 MYH7 NM_000257 BIN1 NM_139343 MYOT NM_006790 BVES NM_007073 NEB NM_004543 CAPN3 NM_000070 PLEC NM_000445 CAV3 NM_033337 POMGNT1 NM_017739 CAVIN1 NM_012232 POMGNT2 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetic Heterogeneity of Motor Neuropathies
Genetic heterogeneity of motor neuropathies Boglarka Bansagi, MD ABSTRACT Helen Griffin, PhD Objective: To study the prevalence, molecular cause, and clinical presentation of hereditary motor Roger G. Whittaker, MD, neuropathies in a large cohort of patients from the North of England. PhD Methods: Detailed neurologic and electrophysiologic assessments and next-generation panel Thalia Antoniadi, PhD testing or whole exome sequencing were performed in 105 patients with clinical symptoms of Teresinha Evangelista, distal hereditary motor neuropathy (dHMN, 64 patients), axonal motor neuropathy (motor MD Charcot-Marie-Tooth disease [CMT2], 16 patients), or complex neurologic disease predominantly James Miller, MD, PhD affecting the motor nerves (hereditary motor neuropathy plus, 25 patients). Mark Greenslade, PhD Natalie Forester, PhD Results: The prevalence of dHMN is 2.14 affected individuals per 100,000 inhabitants (95% – Jennifer Duff, PhD confidence interval 1.62 2.66) in the North of England. Causative mutations were identified in Anna Bradshaw 26 out of 73 index patients (35.6%). The diagnostic rate in the dHMN subgroup was 32.5%, Stephanie Kleinle, PhD which is higher than previously reported (20%). We detected a significant defect of neuromus- Veronika Boczonadi, PhD cular transmission in 7 cases and identified potentially causative mutations in 4 patients with Hannah Steele, MD multifocal demyelinating motor neuropathy. Venkateswaran Ramesh, Conclusions: Many of the genes were shared between dHMN and motor CMT2, indicating identical MD disease mechanisms; therefore, we suggest changing the classification and including dHMN also as Edit Franko, MD, PhD a subcategory of Charcot-Marie-Tooth disease. Abnormal neuromuscular transmission in some Angela Pyle, PhD genetic forms provides a treatable target to develop therapies. -

Epistasis-Driven Identification of SLC25A51 As a Regulator of Human
ARTICLE https://doi.org/10.1038/s41467-020-19871-x OPEN Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import Enrico Girardi 1, Gennaro Agrimi 2, Ulrich Goldmann 1, Giuseppe Fiume1, Sabrina Lindinger1, Vitaly Sedlyarov1, Ismet Srndic1, Bettina Gürtl1, Benedikt Agerer 1, Felix Kartnig1, Pasquale Scarcia 2, Maria Antonietta Di Noia2, Eva Liñeiro1, Manuele Rebsamen1, Tabea Wiedmer 1, Andreas Bergthaler1, ✉ Luigi Palmieri2,3 & Giulio Superti-Furga 1,4 1234567890():,; About a thousand genes in the human genome encode for membrane transporters. Among these, several solute carrier proteins (SLCs), representing the largest group of transporters, are still orphan and lack functional characterization. We reasoned that assessing genetic interactions among SLCs may be an efficient way to obtain functional information allowing their deorphanization. Here we describe a network of strong genetic interactions indicating a contribution to mitochondrial respiration and redox metabolism for SLC25A51/MCART1, an uncharacterized member of the SLC25 family of transporters. Through a combination of metabolomics, genomics and genetics approaches, we demonstrate a role for SLC25A51 as enabler of mitochondrial import of NAD, showcasing the potential of genetic interaction- driven functional gene deorphanization. 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Laboratory of Biochemistry and Molecular Biology, Department of Biosciences, Biotechnologies and Biopharmaceutics, -

Comparison of Human Solute Carriers
Comparison of human solute carriers Avner Schlessinger,1,2,3* Pa¨ r Matsson,1,2,3 James E. Shima,1,2,3,4 Ursula Pieper,1,2,3 Sook Wah Yee,1,2,3 Libusha Kelly,1,2,3,5 Leonard Apeltsin,1,2,3,5 Robert M. Stroud,6 Thomas E. Ferrin,1,2,3 Kathleen M. Giacomini,1,2,3 and Andrej Sali1,2,3* 1Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, California 2Department of Pharmaceutical Chemistry, University of California, San Francisco, California 3California Institute for Quantitative Biosciences, University of California, San Francisco, California 4Graduate Program in Pharmaceutical Sciences and Pharmacogenomics, University of California, San Francisco, California 5Graduate Program in Biological and Medical Informatics, University of California, San Francisco, California 6Department of Biochemistry and Biophysics, University of California, San Francisco, California Received 14 August 2009; Revised 10 December 2009; Accepted 14 December 2009 DOI: 10.1002/pro.320 Published online 5 January 2010 proteinscience.org Abstract: Solute carriers are eukaryotic membrane proteins that control the uptake and efflux of solutes, including essential cellular compounds, environmental toxins, and therapeutic drugs. Solute carriers can share similar structural features despite weak sequence similarities. Identification of sequence relationships among solute carriers is needed to enhance our ability to model individual carriers and to elucidate the molecular mechanisms of their substrate specificity and transport. Here, we describe a comprehensive comparison of solute carriers. We link the proteins using sensitive profile–profile alignments and two classification approaches, including similarity networks. The clusters are analyzed in view of substrate type, transport mode, organism conservation, and tissue specificity. -

Frontiersin.Org 1 April 2015 | Volume 9 | Article 123 Saunders Et Al
ORIGINAL RESEARCH published: 28 April 2015 doi: 10.3389/fnins.2015.00123 Influx mechanisms in the embryonic and adult rat choroid plexus: a transcriptome study Norman R. Saunders 1*, Katarzyna M. Dziegielewska 1, Kjeld Møllgård 2, Mark D. Habgood 1, Matthew J. Wakefield 3, Helen Lindsay 4, Nathalie Stratzielle 5, Jean-Francois Ghersi-Egea 5 and Shane A. Liddelow 1, 6 1 Department of Pharmacology and Therapeutics, University of Melbourne, Parkville, VIC, Australia, 2 Department of Cellular and Molecular Medicine, University of Copenhagen, Copenhagen, Denmark, 3 Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia, 4 Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland, 5 Lyon Neuroscience Research Center, INSERM U1028, Centre National de la Recherche Scientifique UMR5292, Université Lyon 1, Lyon, France, 6 Department of Neurobiology, Stanford University, Stanford, CA, USA The transcriptome of embryonic and adult rat lateral ventricular choroid plexus, using a combination of RNA-Sequencing and microarray data, was analyzed by functional groups of influx transporters, particularly solute carrier (SLC) transporters. RNA-Seq Edited by: Joana A. Palha, was performed at embryonic day (E) 15 and adult with additional data obtained at University of Minho, Portugal intermediate ages from microarray analysis. The largest represented functional group Reviewed by: in the embryo was amino acid transporters (twelve) with expression levels 2–98 times Fernanda Marques, University of Minho, Portugal greater than in the adult. In contrast, in the adult only six amino acid transporters Hanspeter Herzel, were up-regulated compared to the embryo and at more modest enrichment levels Humboldt University, Germany (<5-fold enrichment above E15). -

Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: a Review
1 SUPPLEMENTARY MATERIAL TO: Diseases caused by mutations in mitochondrial carrier genes SLC25: a review Authors: Ferdinando Palmieri1,*, Pasquale Scarcia1 and Magnus Monné1,2,* Affiliations: 1Department of Biosciences, Biotechnologies and Biopharmaceutics, Laboratory of Biochemistry and Molecular Biology, University of Bari Aldo Moro, Via E. Orabona 4, 70125 Bari, Italy, 2Department of Sciences, University of Basilicata, Via Ateneo Lucano 10, 85100 Potenza, Italy *Corresponding authors: F. Palmieri (Department of Biosciences, Biotechnologies and Biopharmaceutics, University of Bari, Via E. Orabona 4, 70125 Bari, Italy, 0039- 0805443323, [email protected]) and M. Monné (Department of Sciences, University of Basilicata, Via Ateneo Lucano 10, 85100 Potenza, Italy, [email protected]). Table S1. Disease-causing mutations in mitochondrial carriers. For each gene/carrier the mutations are listed in the following order: first the nonsense, deletion/insertion and splicing mutations, and afterwards the missense mutations. Mitochondrial Disease-associated Mutation effect in Homo- / References carrier mutation the protein (or Hetero- problem in zygous splicing) patients SLC25A1, CIC c.18_24dup p.Ala9Profs*82 2/1 (1) (2) c.517_526del p.Arg173Glyfs*2 1/1 (1) (3) c.648_655del p.Met218Serfs*25 0/1 (3) c.768C > G p.Tyr256* 0/1 (1) Position in c.821C > T p.Ala274Ilefs*24 2/4 (1) Fig. 2 10 c.82G > A p.Ala28Thr 0/1 (3) 22 c.119T > A p.Ile40Asn 0/1 (3) 27 c.134C > T p.Pro45Leu 0/2 (1)(2) 29 c.139G > A p.Glu47Lys 1/0 (3) 55 c.205G > T p.Asp69Tyr -

The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology
University of Groningen The SLC25 Carrier Family Kunji, Edmund R S; King, Martin S; Ruprecht, Jonathan J; Thangaratnarajah, Chancievan Published in: Physiology DOI: 10.1152/physiol.00009.2020 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2020 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Kunji, E. R. S., King, M. S., Ruprecht, J. J., & Thangaratnarajah, C. (2020). The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology, 35(5), 302-327. https://doi.org/10.1152/physiol.00009.2020 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. -

RNA-Seq Reveals Conservation of Function Among the Yolk Sacs Of
RNA-seq reveals conservation of function among the PNAS PLUS yolk sacs of human, mouse, and chicken Tereza Cindrova-Daviesa, Eric Jauniauxb, Michael G. Elliota,c, Sungsam Gongd,e, Graham J. Burtona,1, and D. Stephen Charnock-Jonesa,d,e,1,2 aCentre for Trophoblast Research, Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, CB2 3EG, United Kingdom; bElizabeth Garret Anderson Institute for Women’s Health, Faculty of Population Health Sciences, University College London, London, WC1E 6BT, United Kingdom; cSt. John’s College, University of Cambridge, Cambridge, CB2 1TP, United Kingdom; dDepartment of Obstetrics and Gynaecology, University of Cambridge, Cambridge, CB2 0SW, United Kingdom; and eNational Institute for Health Research, Cambridge Comprehensive Biomedical Research Centre, Cambridge, CB2 0QQ, United Kingdom Edited by R. Michael Roberts, University of Missouri-Columbia, Columbia, MO, and approved May 5, 2017 (received for review February 14, 2017) The yolk sac is phylogenetically the oldest of the extraembryonic yolk sac plays a critical role during organogenesis (3–5, 8–10), membranes. The human embryo retains a yolk sac, which goes there are limited data to support this claim. Obtaining experi- through primary and secondary phases of development, but its mental data for the human is impossible for ethical reasons, and importance is controversial. Although it is known to synthesize thus we adopted an alternative strategy. Here, we report RNA proteins, its transport functions are widely considered vestigial. sequencing (RNA-seq) data derived from human and murine yolk Here, we report RNA-sequencing (RNA-seq) data for the human sacs and compare them with published data from the yolk sac of and murine yolk sacs and compare those data with data for the the chicken. -
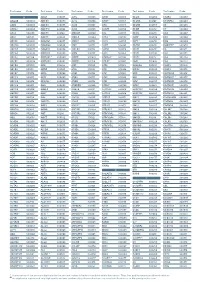
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -

Integrated Identification of Key Genes and Pathways in Alzheimer's Disease Via Comprehensive Bioinformatical Analyses
Yan et al. Hereditas (2019) 156:25 https://doi.org/10.1186/s41065-019-0101-0 RESEARCH Open Access Integrated identification of key genes and pathways in Alzheimer’s disease via comprehensive bioinformatical analyses Tingting Yan* , Feng Ding and Yan Zhao Abstract Background: Alzheimer’s disease (AD) is known to be caused by multiple factors, meanwhile the pathogenic mechanism and development of AD associate closely with genetic factors. Existing understanding of the molecular mechanisms underlying AD remains incomplete. Methods: Gene expression data (GSE48350) derived from post-modern brain was extracted from the Gene Expression Omnibus (GEO) database. The differentially expressed genes (DEGs) were derived from hippocampus and entorhinal cortex regions between AD patients and healthy controls and detected via Morpheus. Functional enrichment analyses, including Gene Ontology (GO) and pathway analyses of DEGs, were performed via Cytoscape and followed by the construction of protein-protein interaction (PPI) network. Hub proteins were screened using the criteria: nodes degree≥10 (for hippocampus tissues) and ≥ 8 (for entorhinal cortex tissues). Molecular Complex Detection (MCODE) was used to filtrate the important clusters. University of California Santa Cruz (UCSC) and the database of RNA-binding protein specificities (RBPDB) were employed to identify the RNA-binding proteins of the long non-coding RNA (lncRNA). Results: 251 & 74 genes were identified as DEGs, which consisted of 56 & 16 up-regulated genes and 195 & 58 down-regulated genes in hippocampus and entorhinal cortex, respectively. Biological analyses demonstrated that the biological processes and pathways related to memory, transmembrane transport, synaptic transmission, neuron survival, drug metabolism, ion homeostasis and signal transduction were enriched in these genes. -

Pdf PMID: 23988125 28
RESEARCH ARTICLE Bovine and murine models highlight novel roles for SLC25A46 in mitochondrial dynamics and metabolism, with implications for human and animal health Amandine Duchesne1*, Anne Vaiman1, Johan Castille1, Christian Beauvallet1, Pauline Gaignard2, Sandrine Floriot1, Sabrina Rodriguez1,3, Marthe Vilotte1, Laurent Boulanger4, Bruno Passet1, Olivier Albaric5, FrancËois Guillaume1, a1111111111 Abdelhak Boukadiri1, Laurence Richard6, Maud Bertaud1, Edouard Timsit7, a1111111111 RaphaeÈl Guatteo8, Florence JaffreÂzic1, Pierre Calvel1, Louise Helary1, Rachid Mahla9, a1111111111 Diane EsquerreÂ10, Christine PeÂchoux1, Sophie Liuu11, Jean-Michel Vallat6, a1111111111 Didier Boichard1, Abdelhamid Slama2, Jean-Luc Vilotte1 a1111111111 1 GABI, INRA, AgroParisTech, Universite Paris-Saclay, Jouy-en-Josas, France, 2 Biochemistry Laboratory, Bicêtre Hospital, Assistance-Publique HoÃpitaux de Paris, University Paris-Sud, Le Kremlin-Bicêtre Cedex, France, 3 TWB, Universite de Toulouse, INRA, INSA, CNRS, Ramonville Saint-Agne, France, 4 UMR BDR, INRA, ENVA, Universite Paris Saclay, Jouy en Josas, France, 5 LHA, Oniris, Nantes Atlantic College of Veterinary Medecine, Food Science and Engineering, Universite Nantes Angers Le Mans, Nantes, France, OPEN ACCESS 6 Department of Neurology, National Reference Center for Rare Peripheral Neuropathies, University Hospital, Limoges, France, 7 Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada, Citation: Duchesne A, Vaiman A, Castille J, 8 BIOEPAR, INRA, Oniris, Nantes, France, 9 Labogena, Jouy-en-Josas, France, 10 GenPhySE, Universite Beauvallet C, Gaignard P, Floriot S, et al. (2017) de Toulouse, INRA, INPT, ENVT, Castanet Tolosan, France, 11 Micalis Institute, INRA, AgroParisTech, Bovine and murine models highlight novel roles for Universite Paris-Saclay, Jouy-en-Josas, France SLC25A46 in mitochondrial dynamics and metabolism, with implications for human and * [email protected] animal health.