Polystyrene/Poly(Methyl Methacrylate) Blends in the Presence of Cyclohexane: Selective Solvent Washing Or Equilibrium Adsorption?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

EPA 450 3-83-008 Control of VOC Emissions from Manufacture Of
dine Series Emission Standards and Engineering Division Office of ~ir,.~ofp,and Radiation Office of Air Qualify P!anning and Standards Research Triangle Park: .North Carolina 2771 1 November 1 983 I GUIDELINE SERIES I The guideline series of reports is issued by the Office of Air Quality Planning and Standards (OAQPS) to provide information to state and local air pollution control agencies; for example, to provide guidance on the acquisition and processing of air qualitydata and on the planning and analysis requisite for the maintenance of air quality. Reports published in this series will be available - as supplies permit - from the Library Services Office (MD-35), U.S. Environmental Protection Agency, Research Triangle Park, North Carolina 2771 1, orfor a nominal fee, from the National Technical Information Service, 5285 Port Royal Road, Springfield, Virginia 221 61. TABLE OF CONTENTS INTRODUCTION ................ PROCESS,AND POLLUTANT EMISSIONS ..... INTRODUCTION ............ POLYPROPYLENE ............ 2.2.1 General Industry Description . 2.2.2 Model Plant ......... HIGH-DENSITY POLYETHYLENE ...... 2.3 .I General Industry Description . 2.3.2 Model Plant. ......... POLYSTYRENE . 2.4.1 General Industry Description . 2,4,2 Model Plant ....... REFERENCES FOR CHAPTER 2. .... EMISSION CONTROL TECHNIQUES. ..... 3.1 CONTROL BY COMBUSTION TECHNIQUES. 3.1.1 Flares .......... 3.1.2 Thermal Incinerators ... 3.1.3 Catalytic Incinerators . 3.1.4 Industrial Boilers .... 3.2 CONTROL BY RECOVERY TECHNIQUES . 3.2.1 Condensers ........ 3.2.2 Adsorbers ........ 3.2.3 Absorbers ........ 3.3 REFERENCES FOR CHAPTER 3. .... ENVIRONMENTAL ANALYSIS OF RACT .... 4.1 INTRODUCTION. .......... 4.2 AIR POLLUTION .......... 4.3 WATER POLLUTION ......... 4.4 SOLID WASTE DISPOSAL. -

Research Article Preparation, Properties, and Self-Assembly Behavior of PTFE-Based Core-Shell Nanospheres
Hindawi Publishing Corporation Journal of Nanomaterials Volume 2012, Article ID 980541, 15 pages doi:10.1155/2012/980541 Research Article Preparation, Properties, and Self-Assembly Behavior of PTFE-Based Core-Shell Nanospheres Katia Sparnacci,1 Diego Antonioli,1 Simone Deregibus,1 Michele Laus,1 Giampaolo Zuccheri,2 Luca Boarino,3 Natascia De Leo,3 and Davide Comoretto4 1 Dipartimento di Scienze dell’ Ambiente e della Vita, Universita` del Piemonte Orientale “A. Avogadro”, INSTM, UdR Alessandria, Via G. Bellini 25 g, 15100 Alessandria, Italy 2 Dipartimento di Biochimica “G. Moruzzi”, Universita` di Bologna, INSTM, CNRNANO-S3, Via Irnerio 48, 40126 Bologna, Italy 3 NanoFacility Piemonte, Electromagnetism Division, Istituto Nazionale di Ricerca Metrologica Strada delle Cacce 91, 10135 Torino, Italy 4 Dipartimento di Chimica e Chimica Industriale, Universita` degli Studi di Genova, Via Dodecaneso 31, 16146 Genova, Italy Correspondence should be addressed to Michele Laus, [email protected] Received 2 August 2011; Revised 17 October 2011; Accepted 24 October 2011 Academic Editor: Hai-Sheng Qian Copyright © 2012 Katia Sparnacci et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Nanosized PTFE-based core-shell particles can be prepared by emulsifier-free seed emulsion polymerization technique starting from spherical or rod-like PTFE seeds of different size. The shell can be constituted by the relatively high Tg polystyrene and polymethylmethacrylate as well as by low Tg polyacrylic copolymers. Peculiar thermal behavior of the PTFE component is observed due to the high degree of PTFE compartmentalization. -

A Summary of the NBS Literature Reviews on the Chemical Nature And
r NATL INST. OF STAND & TECH NBS Reference PUBLICATIONS 1 AlllDS Tfi37fiE NBSIR 85-326tL^ A Summary of the NBS Literature Reviews on the Chemical Nature and Toxicity of the Pyrolysis and Combustion Products from Seven Plastics: Acrylonitrile- Butadiene- Styrenes (ABS), Nylons, Polyesters, Polyethylenes, Polystyrenes, Poly(Vinyl Chlorides) and Rigid Polyurethane Foams Barbara C. Levin U.S. DEPARTMENT OF COMMERCE National Bureau of Standards National Engineering Laboratory Center for Fire Research Gaithersburg, MD 20899 June 1986 Sponsored in part by: g Consumer Product Safety Commission sda. MD 20207 100 .056 85-3267 1986 4 NBS RESEARCH INFORf/ATION CENTER NBSIR 85-3267 A SUMMARY OF THE NBS LITERATURE REVIEWS ON THE CHEMICAL NATURE AND TOXICITY OF THE PYROLYSIS AND I COMBUSTION PRODUCTS FROM SEVEN PLASTICS: ACRYLONITRILE-BUTADIENE- STYRENES (ABS), NYLONS, POLYESTERS, POLYETHYLENES, POLYSTYRENES, POLY(VINYL CHLORIDES) AND RIGID POLYURETHANE FOAMS Barbara C. Levin U.S. DEPARTMENT OF COMMERCE National Bureau of Standards National Engineering Laboratory Center for Fire Research Gaithersburg, MD 20899 June 1 986 Sponsored in part by: The U.S. Consumer Product Safety Commission Bethesda, MD 20207 U.S. DEPARTMENT OF COMMERCE, Malcolm Baldrige, Secretary NATIONAL BUREAU OF STANDARDS. Ernest Ambler. Director Table of Contents Page Abstract 1 1.0 Introduction 2 2.0 Scope 3 3.0 Thermal Decomposition Products 4 4.0 Toxicity 9 5.0 Conclusion 13 6.0 Acknowledgements 14 References 15 iii List of Tables Page Table 1. Results of Bibliographic Search 17 Table 2 . Thermal Degradation Products 18 Table 3. Test Methods Used to Assess Toxicity of the Thermal Decomposition Products of Seven Plastics 26 Table 4. -

Synthesis and Characterization of Functionalized Poly(Arylene Ether Sulfone)S Using Click Chemistry
Wright State University CORE Scholar Browse all Theses and Dissertations Theses and Dissertations 2016 Synthesis and Characterization of Functionalized Poly(arylene ether sulfone)s using Click Chemistry Kavitha Neithikunta Wright State University Follow this and additional works at: https://corescholar.libraries.wright.edu/etd_all Part of the Chemistry Commons Repository Citation Neithikunta, Kavitha, "Synthesis and Characterization of Functionalized Poly(arylene ether sulfone)s using Click Chemistry" (2016). Browse all Theses and Dissertations. 1650. https://corescholar.libraries.wright.edu/etd_all/1650 This Thesis is brought to you for free and open access by the Theses and Dissertations at CORE Scholar. It has been accepted for inclusion in Browse all Theses and Dissertations by an authorized administrator of CORE Scholar. For more information, please contact [email protected]. SYNTHESIS AND CHARACTERIZATION OF FUNCTIONALIZED POLY (ARYLENE ETHER SULFONE)S USING CLICK CHEMSITRY A thesis submitted in partial fulfilment of the requirements for the degree of Master of Science By Kavitha Neithikunta B.sc Osmania University, 2010 2016 Wright State University WRIGHT STATE UNIVERSITY GRADUATE SCHOOL August 26, 2016 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MYSUPERVISION BY Kavitha Neithikunta ENTITLED Synthesis and Characterization of Functionalized Poly(arylene ether sulfone)s using Click chemistry BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science __________________________ Eric Fossum, Ph.D. Thesis Advisor ___________________________ David Grossie, Ph.D. Chair, Department of Chemistry Committee on Final Examination ____________________________ Eric Fossum, Ph.D. _____________________________ Daniel M. Ketcha, Ph.D. _____________________________ William A. Feld, Ph.D. _______________________________ Robert E. W. Fyffe, Ph.D Vice President for Research and Dean of the Graduate School ABSTRACT Neithikunta, Kavitha M.S., Department of Chemistry, Wright State University, 2016. -

Optical Properties of Poly (Vinyl Chloride)/Polystyrene Blends
International Journal of Application or Innovation in Engineering & Management (IJAIEM) Web Site: www.ijaiem.org Email: [email protected] Volume 3, Issue 5, May 2014 ISSN 2319 - 4847 Optical properties of Poly (Vinyl chloride)/polystyrene blends Asrar Abdulmunem Saeed & Mohammed Zorah Hassan Mustansiriyah University- College of Science, Baghdad, Iraq Abstract Poly (Vinyl chloride) (PVC)/polystyrene (PS) blends with different ratios were prepared by solvent casting from tetrahydrofuran (THF). The absorption spectra of polyvinylchloride / polystyrene blends at different concentrations (PVC %, 75% PVC/25% PS, 50% PVC / 50%PS, 25% PVC/75% PS, % PS) showed absorption changes in the wavelength range, which depends on the polymer type, and on the concentration of the polymer blends. It was found that 50 % PVC / 50 % PS ratio from these polymers showed higher absorption values in comparison with the other blends. The absorption spectra has been recorded in the wave length range (200 –1100) nm. The absorption coefficients (α), the extinction coefficient (K), refractive index (n) have been evaluated. Key words: optical properties, PVC/PS blend, optical constants. 1- Introduction Polymer blends are a mixture of chemically different polymers or copolymers with no covalent bonding between them. Polymer blends are classified as three types, namely, homologous, miscible and immiscible blends. Chemically identical polymers with different molecular masses constitute homologous polymer lends. Miscible polymer blends exhibit single phase behavior and immiscible polymer blends exhibit two or more phases at all compositions and temperatures. Preparation of polymer blends has been received considerable importance in the recent past owing to the shorter time and lower cost of the product development than those of a new polymer [1]. -

Monomers – Styrene and Vinyl Chloride
Chemical Information Sheet Version 2.0 | March 2021 MONOMERS – STYRENE AND VINYL CHLORIDE Other Names Styrene: Ethenylbenzene, vinylbenzene, Monomers are chemical precursors that link together to phenylethene create polymer materials. Styrene and vinyl chloride are Vinyl Chloride: VCM, chloroethene monomers that may be present in low concentrations in some polymer materials. The presence of these monomers can be related to the process controls during CAS Number Substance polymer production. 100-42-5 Styrene Uses in the Supply Chain 75-01-4 Vinyl Chloride Styrene is a colorless liquid that evaporates easily which may be used to create polymers including polystyrene, ABS plastic, May Be Found In Styrene: Polystyrene, Acrylonitrile-butadiene- synthetic rubber (SBR) and other materials. Styrene can also be styrene (ABS) plastic, Styrene-butadiene rubber (SBR), styrene-divinylbenzene (S-DVB) used in plastic packaging and electrical parts. Vinyl Chloride: Polyvinyl chloride (PVC), Vinyl Chloride is used in production of polyvinyl chloride vinyl polymers, plastisol screen prints, plastic (PVC) and vinyl polymers, which can be hard or flexible parts, coatings for leather, synthetic leather and materials. PVC can be associated with plastisol screen prints, textiles plastic parts, and a variety of coatings on leather, synthetic leather, and textiles. Why Monomers Are Restricted ▪ Legislation in major markets globally restricts or regulates the presence of styrene and vinyl chloride in finished products or materials. ▪ Monomers can present a variety -

Plastics What Are They?
Plastics What are they? Plastic What is it? A synthetic or semi-synthetic material made of organic polymers. A polymer is a molecule of many monomers bonded together. • Organic means containing carbon. • Monomer means likes to bond with others like it. • Polymer means a chain of monomers. • Plastic becomes many chains together, some linked together and some not linked together. Plastics Classification Plastics consist of molecules called monomers that like to form long chains. These long chains can be connected to one another to varying degrees. The connections between chains are called cross-links. The fewer cross links, the more likely the plastic can be recycled. Thermoplastics Elastomers Thermosets Little to no Cross-Linking Some cross-linking Extensive cross-linking Plastics Basic Classification Thermoplastics Little to no Cross-Linking • Become pliable and moldable when heated. • Solidify when cooled. • Have a wide range of applications (e.g. food packaging, bumpers, credit cards). • Can be recycled. • Can be reused as filler material for lightweight concrete, composite deck boards, etc. Plastics Basic Classification Elastomers Some cross-linking • Are composed of long polymer chains that reconfigure under applied stress. • Become non-rigid solids above their “glass transition temperatures”. • Can be stretched repeatedly and returned to their original shape. • Are also referred to as rubber. • Are predominantly thermoset plastics and cannot be recycled. Plastics In 1988, the Society of the Plastics Industry (SPI) developed a coding -

Polymer Properties and Classification
Polymer Characterization LDPE Polyethylene low density HDPE Polyethylene high density ABS Acrylonitrile-butadiene-styrene SAN Styrene-acrylonitrile copolymer EVA Polyethylene co-vinyl acetate PVA Polyvinyl acetate PerkinElmer Solutions for Polymer Characterization Tg(ºC): -130 to 100 Cp (J/g*K): 1,8 to 3,4 Tg(ºC): -130 to 100 Cp (J/g*K): 1,8 to 3,4 Tg(ºC): 110 to 125 CpJ/(g*K): 1,25 to 1,7 Tg(ºC): 95 to 110 CpJ/(g*K): 1,2 Tg(ºC): -45 to 20 CpJ/(g*K): 2,3 Tg(ºC): 25 to 35 CpJ/(g*K): - Tm(ºC): 100 to 120 DHf (J/g): - Tm(ºC): 130 to 140 DHf (J/g): 293 Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm(ºC): 30 to 100 DHf (J/g): 10 to 100 Tm( ºC ): - DHf (J/g): - Td(ºC): 490 to 500 Td(ºC): 490 to 500 Td(ºC): 420 Td(ºC): 420 Td(ºC): 480 Td(ºC): - PP Polypropylene PS PMMA Polymethylmethacrylate PBMA CA Polystyrene Polybuthylmethacrylate Cellulose acetate EP Epoxy resin Molecular Spectroscopy FTIR Differential Scanning Calorimetry Tg(ºC): -20 to -5 CpJ/(g*K): 1,8 Tg(ºC): 90 to 110 Cp (J/g*K): 1,8 to 3,4 Tg(ºC): 85 to 100 CpJ/(g*K): 1,45 to 1,5 Tg(ºC): 15 to 25 CpJ/(g*K): - Tg(ºC): 45 to 60 CpJ/(g*K): - Tg(ºC): 50 to 200 CpJ/(g*K): 1,6 to 2,1 Identify and quantitate organic molecules and compounds, Glass transition & melting temperatures, crystallinity, heat of Understand chemical & physical composition of laminates & fusion, reaction rates, specific heat & heat capacity, curing, Tm(ºC): 165 to 175 DHf (J/g): 207 Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm( ºC): - DHf (J/g): - adhesives , Troubleshoot -

Styrofoam—Expanded Polystyrene
Styrofoam—Expanded Polystyrene Polystyrene (PS #6) is the most widely used form of plastic in consumer goods. California produces 3.32 billion pounds of polystyrene every year for packaging and food containers. Rarely recycled and used for only minutes, this throw-away plastic has a long-lasting impact on our environment and on our health. Polystyrene is found in two forms—expanded polystyrene (EPS, or trademark name Styrofoam™), and extruded polystyrene, which is a clear, rigid plastic used in food clamshells and CD cases. Neither of these plastics can be effectively recycled, despite the #6 and small recycling symbol that appears on the bottom of the package. Intact or broken down bits of foam polystyrene, particularly from food packaging, are some of the most commonly found plastic items on beaches and inland creeks. Expanded polystyrene litter has been such a problem that at least 40 cities and counties in California have either banned EPS food packaging or require recyclable single-use food packaging. Marine Life Impacts Like other plastics, EPS does not biodegrade in the marine environment and may persist for hundreds of years. EPS is lightweight and buoyant, properties that make it easily carried by water and wind to the ocean via storm drains. Foam products readily break apart into smaller pieces that are commonly mistaken for food by marine life. When animals eat EPS and other plastics, their digestive systems get clogged, preventing them from absorbing nutrients and ultimately leading to starvation. Surface-feeding species of birds like petrels commonly ingest this floating plastic, and jellyfish and other marine life that filter water for nutrients are becoming increasingly “plasticized” as the ocean fills microscopic pieces of plastic. -
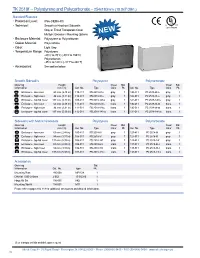
TK 2518F – Polystyrene and Polycarbonate – 254X180mm (10.0X7.09In.)
TK 2518f – Polystyrene and Polycarbonate – 254x180mm (10.0x7.09in.) Standard Features • Protection Level: IP66 (NEMA 4X) • Technical: Smooth or Knockout Sidewalls Upon Request Gray or Tinted Transparent Cover ➀ Multiple Enclosure Mounting Options NEW • Enclosure Material: Polystyrene or Polycarbonate • Gasket Material: Polyurethane • Color: Light Gray • Temperature Range: Polystyrene -40°C to 70°C (-40°F to 158°F) Polycarbonate -35°C to 120°C (-31°F to 248°F) • Accessories: See section below Smooth Sidewalls Polystyrene Polycarbonate Ordering Height Cover Std. Cover Std. Information mm (in) Cat. No. Type Color Pk. Cat. No. Type Color Pk. Enclosure - low cover 63 mm (2.48 in) 110-411 PS 2518-6f-o gray 1 120-411 PC 2518-6f-o gray 1 Enclosure - high cover 84 mm (3.31 in) 110-911 PS 2518-8f-o gray 1 120-911 PC 2518-8f-o gray 1 Enclosure - top hat cover 137 mm (5.39 in) 109-411 PS 2518-14f-o gray 1 121-411 PC 2518-14f-o gray 1 Enclosure - low cover 63 mm (2.48 in) 111-011 PS 2518-6f-to trans. 1 130-011 PC 2518-6f-to trans. 1 Enclosure - high cover 84 mm (3.31 in) 111-511 PS 2518-8f-to trans. 1 130-511 PC 2518-8f-to trans. 1 Enclosure - top hat cover 137 mm (5.39 in) 112-011 PS 2518-14f-to trans. 1 131-011 PC 2518-14f-to trans. 1 Sidewalls with Metric Knockouts Polystyrene Polycarbonate Ordering Height Cover Std. Cover Std. Information mm (in) Cat. No. Type Color Pk. -

B-Polystyrene Compatibilizer on the Recycled Polypropylene And
Received: 26 February 2018 Revised: 28 April 2018 Accepted: 28 April 2018 DOI: 10.1002/pat.4346 RESEARCH ARTICLE Effects of polystyrene‐b‐poly(ethylene/propylene)‐b‐ polystyrene compatibilizer on the recycled polypropylene and recycled high‐impact polystyrene blends Yufei Kong1 | Yingchun Li1 | Guosheng Hu1 | Nuo Cao2 | Youquan Ling1 | Duo Pan3,4 | Qian Shao4 | Zhanhu Guo3 1 School of Materials Science and Engineering, North University of China, Taiyuan 030051, The recycled polypropylene/recycled high‐impact polystyrene (R‐PP/R‐HIPS) blends China were melt extruded by twin‐screw extruder and produced by injection molding 2 China National Electric Apparatus Research ‐ ‐ ‐ ‐ Institute Co, Ltd, Guangzhou 510000, China machine. The effects of polystyrene b poly(ethylene/propylene) b polystyrene copol- 3 Department of Chemical and Biomolecular ymer (SEPS) used as compatibilizer on the mechanical properties, morphology, melt Engineering, University of Tennessee, flow index, equilibrium torque, and glass transition temperature (Tg) of the blends Knoxville, TN 37996, USA were investigated. It was found that the notch impact strength and the elongation 4 College of Chemical and Environmental Engineering, Shandong University of Science at break of the R‐PP/R‐HIPS blends with the addition of 10 wt% SEPS were and Technology, Qingdao 266590, China 6.46 kJ/m2 and 31.96%, which were significantly improved by 162.46% and Correspondence Yingchun Li, School of Materials Science and 57.06%, respectively, than that of the uncompatibilized blends. Moreover, the addi- Engineering, North University of China, tion of SEPS had a negligible effect on the tensile strength of the R‐PP/R‐HIPS Taiyuan 030051, China. -

Recent Developments in Recycling of Polystyrene Based Plastics
Accepted Manuscript Recent developments in recycling of polystyrene based plastics Sourbh Thakur, Ankit Verma, Bhawna Sharma, Jyoti Chaudhary, Sigitas Tamulevicius, Vijay Kumar Thakur PII: S2452-2236(18)30024-5 DOI: 10.1016/j.cogsc.2018.03.011 Reference: COGSC 136 To appear in: Current Opinion in Green and Sustainable Chemistry Received Date: 1 February 2018 Revised Date: 16 March 2018 Accepted Date: 22 March 2018 Please cite this article as: S. Thakur, A. Verma, B. Sharma, J. Chaudhary, S. Tamulevicius, V.K. Thakur, Recent developments in recycling of polystyrene based plastics, Current Opinion in Green and Sustainable Chemistry (2018), doi: 10.1016/j.cogsc.2018.03.011. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT Recent developments in recycling of polystyrene based plastics Sourbh Thakur a,b* , Ankit Verma b, Bhawna Sharma b, Jyoti Chaudhary b, Sigitas Tamulevicius a, Vijay Kumar Thakur c* aInstitute of Materials Science of Kaunas University of Technology, Barsausko 59, LT-51423 Kaunas, Lithuania bSchool of Chemistry, Shoolini University, Solan 173212, Himachal Pradesh, India cEnhanced Composites and Structures Center, School of Aerospace, Transport and Manufacturing, Cranfield University, Bedfordshire MK43 0AL, UK Abstract Due to their superior properties, plastics derived from petroleum have been extensively used almost in everyday life since last few decades.