Comparative Transcriptome Analysis of Non-Model Taxus Species for Taxol Biosynthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Department of Planning and Zoning
Department of Planning and Zoning Subject: Howard County Landscape Manual Updates: Recommended Street Tree List (Appendix B) and Recommended Plant List (Appendix C) - Effective July 1, 2010 To: DLD Review Staff Homebuilders Committee From: Kent Sheubrooks, Acting Chief Division of Land Development Date: July 1, 2010 Purpose: The purpose of this policy memorandum is to update the Recommended Plant Lists presently contained in the Landscape Manual. The plant lists were created for the first edition of the Manual in 1993 before information was available about invasive qualities of certain recommended plants contained in those lists (Norway Maple, Bradford Pear, etc.). Additionally, diseases and pests have made some other plants undesirable (Ash, Austrian Pine, etc.). The Howard County General Plan 2000 and subsequent environmental and community planning publications such as the Route 1 and Route 40 Manuals and the Green Neighborhood Design Guidelines have promoted the desirability of using native plants in landscape plantings. Therefore, this policy seeks to update the Recommended Plant Lists by identifying invasive plant species and disease or pest ridden plants for their removal and prohibition from further planting in Howard County and to add other available native plants which have desirable characteristics for street tree or general landscape use for inclusion on the Recommended Plant Lists. Please note that a comprehensive review of the street tree and landscape tree lists were conducted for the purpose of this update, however, only -

Cop14 Prop. 36
CoP14 Prop. 36 CONVENTION ON INTERNATIONAL TRADE IN ENDANGERED SPECIES OF WILD FAUNA AND FLORA ____________________ Fourteenth meeting of the Conference of the Parties The Hague (Netherlands), 3-15 June 2007 CONSIDERATION OF PROPOSALS FOR AMENDMENT OF APPENDICES I AND II A. Proposal Amendment of the listing of Taxus cuspidata in Appendix II by: 1. Deleting the phrase "and infraspecific taxa of this species"; and 2. Annotating to read as follows: "Specimens of hybrids and cultivars are not subject to the provisions of the Convention". B. Proponent United States of America C. Supporting statement 1. Taxonomy 1.1 Class: Pinopsida 1.2 Order: Taxales 1.3 Family: Taxaceae 1.4 Genus, species or subspecies, including author and year: Taxus cuspidata Siebold & Zuccarni 1846 1.5 Scientific synonyms: --- 1.6 Common names: English: Japanese yew French: Spanish: 1.7 Code numbers: --- 2. Background The People's Republic of China and the United States of America, in accordance with the consensus recommendation of the 12th meeting of the Plants Committee (Leiden, 2002), prepared a proposal for the 13th meeting of the Conference of the Parties (CoP13; Bangkok, 2004) to include the remaining Asian species of Taxus (i.e., other than T. wallichiana) in Appendix II, but did not propose the inclusion of Taxus spp. from other regions due to the lack of evidence that trade, particularly for the pharmaceutical industry, was adversely affecting species outside of Asia. The proposal for CoP14 Prop. 36 – p. 1 CoP13 therefore only included Taxus chinensis, Taxus cuspidata, Taxus fuana, Taxus sumatrana and all infraspecific taxa of those species (proposal CoP13 Prop. -

The ENEMY: Japanese Yew (Taxus Cuspidata) Strategy: Also Known As the 'Tree of Death' As Dedicated to the Gods of Death. Th
The ENEMY: Japanese yew (Taxus cuspidata) Strategy: Also known as the ‘Tree of Death’ as dedicated to the Gods of Death. This perennial evergreen tree grows to a height of 25 foot if in the right site. This pest is planted all over the U.S. in landscapes and as an ornamental. It is quite often used in Christmas wreaths. Its needles are arranged flat on the stem of which forms red berries. The plant grows in sunny or shady sites as long as there is a medium amount of moisture and well-drained soil. The plant is native to Asia. Due to the types of sites it is planted in it does not generally develop a very deep tap-root. Attack: This plant does not spread very rapid as many other plants do. Its biggest attack on our environment is its toxicity to animals. News stories can be found where 50 antelope were found dead or the elk that were found dead in the Boise foothills. Many livestock producers have had livestock die where they were found to have fed on the Yew tree. Dogs have known to have been killed by eating the stomach of animals that have eaten the tree. Defense: Don’t plant it….In fact due to wildlife issues in the certain areaa, the local officials listed it as a noxious weed and landowners have agreed to remove it from there properties and landscape companies are no longer planting it. If you cut it down ensure that you treat the stump immediately after the cut-down process. -

Reproductive Biology of the Monoecious Clonal Shrub Taxus Canadensis Author(S): Paul Wilson, Michelle Buonopane, Taber D
Torrey Botanical Society Reproductive Biology of the Monoecious Clonal Shrub Taxus canadensis Author(s): Paul Wilson, Michelle Buonopane, Taber D. Allison Reviewed work(s): Source: Bulletin of the Torrey Botanical Club, Vol. 123, No. 1 (Jan. - Mar., 1996), pp. 7-15 Published by: Torrey Botanical Society Stable URL: http://www.jstor.org/stable/2996301 . Accessed: 04/04/2012 14:49 Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at . http://www.jstor.org/page/info/about/policies/terms.jsp JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Torrey Botanical Society is collaborating with JSTOR to digitize, preserve and extend access to Bulletin of the Torrey Botanical Club. http://www.jstor.org Bulletin of the Torrey Botanical Club 123(1), 1996, pp. 7-15 Reproductive biology of the monoecious clonal shrub Taxus canadensis' Paul Wilson,2 Michelle Buonopane3 and Taber D. Allison4 2Department of Biology, California State University, 18111 Nordhoff Street, Northridge, CA 91330-8303 3Department of Biology, Bates College, Lewiston, ME 04240 4Department of Forestry and Wildlife, University of Massachusetts, Amherst, MA 01002 WILSON, P. (Department of Biology, California State University, 18111 Nordhoff Street, Northridge, CA 91330-8303), M. BUONOPANE (Department of Biology, Bates College, Lewiston, ME 04240) AND T D. ALLISON (Department of Forestry and Wildlife, University of Massachusetts, Amherst, MA 01002). -

Taxus (Yew) Production (Draft) by Mark Halcomb UT Area Nursery Specialist
1 Taxus (Yew) Production (draft) by Mark Halcomb UT Area Nursery Specialist Commonly produced yews: Taxus cuspidata 'Capitata' (Capitata Japanese Yew) W type 4, cone upright Taxus cuspidata 'Densa' (Dense Spreading Japanese Yew) H spreader Taxus x media 'Densiformis' (Densiformis Yew) W type 2, semi-spreading Taxus x media 'Hicksii' (Hicks Yew) H, W type 5, broad upright Propagation Yews are propagated by rooted cuttings. Most of our yew producers buy their liners from liner producers rather than root their own. They will be 6-10 inches tall when lined out. Local liner producers are Carl Bouldin, Myers Cove, Phytotektor, Schaefer, Shadow, and Tennessee Valley Nsy in Winchester. Site Selection Yews require a very well drained soil, like dogwood and peach. Select a site without a fragi-pan; where water never stands. Fertility Yews grow best with a soil pH of 6.0--7.0. A medium to high level of phosphorus and potassium is desirable. Soil test early enough so that any lime, phosphate or potash can be broadcast prior to planting. Problem: Yews generally take on a pale green to a yellowish green color during the winter, possibly affecting sales. In September, 1987, Dr. Will Witte, UT Woody Ornamental Researcher with the Experiment Station, shared some Ohio State University research on how they suggest to maintain dark green foliage on yews throughout the winter. The research suggested 6 pounds of actual nitrogen per 1000 square feet or 264 pounds of actual nitrogen per acre per year. It should be equally split into 3-4 applications per year. Four applications would mean 66 pounds actual nitrogen per application. -

Master Plant List
MASTER PLANT LIST 5 N 9 7 8 6 Glasshouse 4 Green Roof 1 2 3 7 MASTER PLANT LIST PAGE 1 TREES 4 Acer griseum PAPERBARK MAPLE 2 3 Acer palmatum ‘Atropurpureum Dissectum’ RED WEEPING CUT-LEAF JAPANESE MAPLE 3 4 5 6 7 Acer palmatum ‘Sango Kaku’ CORAL BARK JAPANESE MAPLE 7 Chamaecyparis nootkatensis ‘Pendula’ WEEPING NOOTKA CYPRESS 7 Chamaecyparis obtusa ‘Gracilis’ SLENDER HINOKI CYPRESS 1 6 Cornus rutgersensis ‘Celestial’ CELESTIAL DOGWOOD 3 6 Davidia involucrata ‘Sonoma’ SONOMA DOVE TREE 4 Gleditsia triacanthos inermis ‘Shademaster’ SHADEMASTER HONEY LOCUST 7 Magnolia grandiflora ‘Teddy Bear’ TEDDY BEAR MAGNOLIA 7 Magnolia grandiflora ‘Bracken’s Brown Beauty’ BRAKEN’S BROWN BEAUTY MAGNOLIA 3 Picea pungens ‘Iseli Fastigiate’ ISELI FASTIGIATE SPRUCE 3 7 Sciadopitys verticillata ‘Wintergreen’ WINTERGREEN UMBRELLA PINE 2 3 Stewartia pseudocamellia JAPANESE STEWARTIA 7 Thuja plicata ‘Atrovirens’ WESTERN RED CEDAR SHRUBS 8 Arbutus compacta DWARF STRAWBERRY TREE 7 Aucuba japonica ‘Rozannie’ ROSANNIE AUCUBA 7 Berberis x gladwynensis ‘William Penn’ BARBERRY 5 Buxus microphylla ‘Wintergreen’ BOXWOOD 8 Callicarpa ‘Profusion’ BEAUTY BERRY 5 7 Camellia sasanqua ‘Yuletide’ YULETIDE CAMELLIA 3 Camellia sasanqua ‘Setsugekka’ SETSUGEKKA CAMELLIA 5 Chaenomeles ‘Dragon’s Blood’ QUINCE 5 Chaenomeles ‘Scarlet Storm’ QUINCE 5 Cornus sericea ‘Bud’s Yellow’ YELLOWTWIG DOGWOOD 1 Corylus avellana ‘Contorta’ HARRY LAUDER’S WALKING STICK 6 Cryptomeria japonica ‘Black Dragon’ BLACK DRAGON JAPANESE CEDAR 8 Cotoneaster dammeri BEARBERRY 2 Daphne genkwa LILAC DAPHNE 4 Dichroa febrifuga CHINESE QUININE 2 Edgeworthia chrysantha ‘Snow Cream’ RICE PAPER SHRUB 7 Fatshedera lizei TREE IVY 7 x Fatshedera lizei ‘Variegata’ VARIGATED TREE IVY 5 Fothergilla gardenii DWARF WITCH ALDER 5 Hamamelis japonica ‘Shibamichi Red’ JAPANESE WITCH HAZEL 2 4 Hydrangea macrophylla ssp. -

Approved Plant List (PDF)
Harford County Approved Plant List April 2019 The Harford County Approved Plant list includes: Permitted tree and plant species for afforestation and reforestation, Permitted street tree species, and Permitted tree and plant species for individual landscaping Please note that not all of the plants on this list are native Maryland species, and therefore not all of the plant species on this list can be used to meet mitigation requirements within the Chesapeake Bay Critical Area. Please refer to the Native Plants list from US Fish and Wildlife, on Harford County’s website, for a comprehensive list of appropriate native plant species to use in the Critical Area. Harford County Approved Plant List April 2019 Plant Species Permitted for Afforestation and Reforestation Trees Scientific Name Common Name Acer negundo Boxelder Acer rubrum Red maple Acer saccharum Sugar maple Acer saccharinum Silver Maple Amelanchier canadensis Shadbush serviceberry Betula lenta Black or Sweet Gum Betula nigra River birch Carpinus caroliniana American hornbeam Carya cordiformis Bitternut hickory Carya glabra Pignut hickory Carya ovata Shagbark hickory Carya tomentosa Mockernut hickory Catalpa speciosa Northern catalpa Celtis occidentalis Common hackberry Cercis canadensis Eastern redbud Cornus florida Flowering dogwood Crataegus crus-galli Cockspur hawthorn Crataegus pruinosa Frosted hawthorn Crataegus punctata Dotted hawthorn Diospyros virginiana Common persimmon Fagus grandifolia American beech Hamamelis virginiana Common witchhazel Juglans cinerea Butternut -

Taxus Wallichiana Var. Mairei 'Jinxishan'
CULTIVAR AND GERMPLASM RELEASES HORTSCIENCE 54(1):181–182. 2019. https://doi.org/10.21273/HORTSCI13252-18 vide more choices for people to use in their gardens. Taxus wallichiana var. mairei Origin ‘Jinxishan’ T. wallichiana var. mairei seeds were collected in the Fall of 1997 from mountain Yalong Qin areas of Fujian Province, China. About Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, 200,000 seeds were selected and sowed in Nanjing 210014, China the Spring of 1999 in Xishan, Wuxi, Jiangsu Province (lat. 31°35#25.09$ N, long. Yiming Chen 120°21#10.54$ E). In 2006, after 7 years in College of Life Sciences, Nanjing Agricultural University, Nanjing, 210095 the juvenile phase, the T. wallichiana var. P.R. China mairei entered its mature phase. Most of the ripe aril on the trees were the usual red color, Weibing Zhuang, Xiaochun Shu, Fengjiao Zhang, and Tao Wang but 11 trees had ripe aril that was yellow. The Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, color of the arils on these trees has remained Nanjing 210014, China stable during the past 9 years. The cultivar name, ‘Jinxishan’, was authorized by the Hui Xu and Bofeng Zhu Forest Variety Certification Committee of Jiangsu Yew Health Technology Co., Ltd., Wuxi 214199, China Jiangsu Province, China, in 2014. Zhong Wang1 Description Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, T. wallichiana var. mairei ‘Jinxishan’ is a Nanjing 210014, China dioecious evergreen tree with average height Additional index words. Taxus wallichiana Zucc., ornamental breeding, mutant of 2.7 m and width of 1.8 m. -

The Reproductive Ecology of the Pacific Yew (Taxus Brevifolia Nutt.) Under a Range of Overstory Conditions in Western Oregon
AN ABSTRACT OF THE THESIS OF Stephen P. DiFazio for the degree of Master of Science in Botany and Plant Pathology presented on May 5, 1995. Title: The Reproductive Ecology of Pacific Yew (Taxus brevifolia Nutt.) Under a Range of Overstory Conditions in Western Oregon Abstract approved:Redacted for Privacy The influence of overstory openness on the reproductive ecology of Pacific yew (Taxus brevifolia Nutt.) was investigated on 4 sites in western Oregon over 2 years. The breeding system of T. brevifolia was found to deviate from pure dioecy under a broad range of canopy and site conditions. Production of female strobili was observed on 17 of 58 predominantly male trees, while no male strobili were observed on 57 female trees. Genet sex ratios were significantly biased in only 1 population, where male genets outnumbered female genets by almost 2 to 1. Mean floral sex ratios were significantly male-biased in all populations and ranged from 5 to 12. Pollen-ovule ratios were in excess of 1,000,000 for all populations. In contrast, reproductive effort based on masses of mature strobili were female-biased by a factor of 1.1 to 5 for all sites. Seed masses also varied inversely with elevation. Pollination phenology varied with elevation and overstory openness. Pollen first began shedding at the lowest sites, and earlier in trees under open conditions than in trees with overstory canopy cover. The duration of pollen shedding varied from 3 to 20 days, and tended to be more protracted at lower sites and under open canopy conditions. Most of the variation in reproductive potential, as indexed by strobilus production, occurred within sites and within trees. -

The Complete Mitochondrial Genome of Taxus Cuspidata (Taxaceae)
Kan et al. BMC Evolutionary Biology (2020) 20:10 https://doi.org/10.1186/s12862-020-1582-1 RESEARCH ARTICLE Open Access The complete mitochondrial genome of Taxus cuspidata (Taxaceae): eight protein- coding genes have transferred to the nuclear genome Sheng-Long Kan1,2, Ting-Ting Shen1, Ping Gong1, Jin-Hua Ran1,2* and Xiao-Quan Wang1,2 Abstract Background: Gymnosperms represent five of the six lineages of seed plants. However, most sequenced plant mitochondrial genomes (mitogenomes) have been generated for angiosperms, whereas mitogenomic sequences have been generated for only six gymnosperms. In particular, complete mitogenomes are available for all major seed plant lineages except Conifer II (non-Pinaceae conifers or Cupressophyta), an important lineage including six families, which impedes a comprehensive understanding of the mitogenomic diversity and evolution in gymnosperms. Results: Here, we report the complete mitogenome of Taxus cuspidata in Conifer II. In comparison with previously released gymnosperm mitogenomes, we found that the mitogenomes of Taxus and Welwitschia have lost many genes individually, whereas all genes were identified in the mitogenomes of Cycas, Ginkgo and Pinaceae. Multiple tRNA genes and introns also have been lost in some lineages of gymnosperms, similar to the pattern observed in angiosperms. In general, gene clusters could be less conserved in gymnosperms than in angiosperms. Moreover, fewer RNA editing sites were identified in the Taxus and Welwitschia mitogenomes than in other mitogenomes, which could be correlated with fewer introns and frequent gene losses in these two species. Conclusions: We have sequenced the Taxus cuspidata mitogenome, and compared it with mitogenomes from the other four gymnosperm lineages. -

National Forest Genetics Laboratory (NFGEL) Species List, August 21, 2013
National Forest Genetics Laboratory (NFGEL) Species List, August 21, 2013 Scientific name Common name Family Order Class Division Kingdom Calocedrus decurrens Incense-cedar Cupressaceae Pinales Pinopsida Coniferophyta Plantae Chamaecyparis lawsoniana Port-Orford cedar Cupressaceae Pinales Pinopsida Coniferophyta Plantae Chamaecyparis nootkatensis Alaskan yellow cedar Cupressaceae Pinales Pinopsida Coniferophyta Plantae Chamaecyparis obtusa Hinoki false cypress Cupressaceae Pinales Pinopsida Coniferophyta Plantae Chamaecyparis thyoides Atlantic white cedar Cupressaceae Pinales Pinopsida Coniferophyta Plantae Cupressocyparis leylandii Leyland cypress Cupressaceae Pinales Pinopsida Coniferophyta Plantae Cupressus bakeri Baker cypress Cupressaceae Pinales Pinopsida Coniferophyta Plantae Cupressus macrocarpa Monterey cypress Cupressaceae Pinales Pinopsida Coniferophyta Plantae Cupressus torulosa Himalayan cypress Cupressaceae Pinales Pinopsida Coniferophyta Plantae Sequoia sempervirens Coast redwood Cupressaceae Pinales Pinopsida Coniferophyta Plantae Sequoiadendron giganteum Giant sequioa Cupressaceae Pinales Pinopsida Coniferophyta Plantae Thuja plicata Western redcedar Cupressaceae Pinales Pinopsida Coniferophyta Plantae Abies concolor White fir Pinaceae Pinales Pinopsida Coniferophyta Plantae Abies fraseri Fraser fir Pinaceae Pinales Pinopsida Coniferophyta Plantae Abies grandis Grand fir Pinaceae Pinales Pinopsida Coniferophyta Plantae Abies lasiocarpa Subalpine fir Pinaceae Pinales Pinopsida Coniferophyta Plantae Abies magnifica Red -
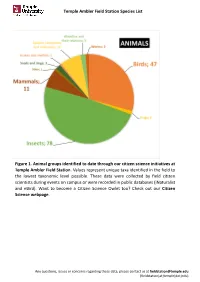
Temple Ambler Field Station Species List Figure 1. Animal Groups Identified to Date Through Our Citizen Science Initiatives at T
Temple Ambler Field Station Species List Figure 1. Animal groups identified to date through our citizen science initiatives at Temple Ambler Field Station. Values represent unique taxa identified in the field to the lowest taxonomic level possible. These data were collected by field citizen scientists during events on campus or were recorded in public databases (iNaturalist and eBird). Want to become a Citizen Science Owlet too? Check out our Citizen Science webpage. Any questions, issues or concerns regarding these data, please contact us at [email protected] (fieldstation[at}temple[dot]edu) Temple Ambler Field Station Species List Figure 2. Plant diversity identified to date in the natural environments and designed gardens of the Temple Ambler Field Station and Ambler Arboretum. These values represent unique taxa identified to the lowest taxonomic level possible. Highlighted are 14 of the 116 flowering plant families present that include 524 taxonomic groups. A full list can be found in our species database. Cultivated specimens in our Greenhouse were not included here. Any questions, issues or concerns regarding these data, please contact us at [email protected] (fieldstation[at}temple[dot]edu) Temple Ambler Field Station Species List database_title Temple Ambler Field Station Species List last_update 22October2020 description This database includes all species identified to their lowest taxonomic level possible in the natural environments and designed gardens on the Temple Ambler campus. These are occurrence records and each taxon is only entered once. This is an occurrence record, not an abundance record. IDs were performed by senior scientists and specialists, as well as citizen scientists visiting campus.