Imaging-Based Pooled CRISPR Screening Reveals Regulators of Lncrna Localization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download PDF (Inglês)
Genetics and Molecular Biology, 44, 1, e20200158 (2021) Copyright © Sociedade Brasileira de Genética. DOI: https://doi.org/10.1590/1678-4685-GMB-2020-0158 Research Article Cellular, Molecular and Developmental Genetics Human DDX56 protein interacts with influenza A virus NS1 protein and stimulates the virus replication 1 2 Ayşegül Pirinçal and Kadir Turan 1Marmara University, Institute of Health Sciences, Istanbul, Turkey. 2Marmara University, Faculty of Pharmacy, Department of Basic Pharmaceutical Sciences, Istanbul, Turkey. Abstract Influenza A viruses (IAV) are enveloped viruses carrying a single-stranded negative-sense RNA genome. Detection of host proteins having a relationship with IAV and revealing of the role of these proteins in the viral replication are of great importance in keeping IAV infections under control. Consequently, the importance of human DDX56, which is determined to be associated with a viral NS1 with a yeast two-hybrid assay, was investigated for IAV replication. The viral replication in knocked down cells for the DDX56 gene was evaluated. The NS1 was co-precipitated with the DDX56 protein in lysates of cells transiently expressing DDX56 and NS1 or infected with the viruses, showing that NS1 and DDX56 interact in mammalian cells. Viral NS1 showed a tendency to co-localize with DDX56 in the cells, transiently expressing both of these proteins, which supports the IP and two-hybrid assays results. The data obtained with in silico predictions supported the in vitro protein interaction results. The viral replication was significantly reduced in the DDX56-knockdown cells comparing with that in the control cells. In conclusion, human DDX56 protein interacts with the IAV NS1 protein in both yeast and mammalian cells and has a positive regulatory effect on IAV replication. -
![DDX56 Mouse Monoclonal Antibody [Clone ID: OTI1G6] Product Data](https://docslib.b-cdn.net/cover/4564/ddx56-mouse-monoclonal-antibody-clone-id-oti1g6-product-data-274564.webp)
DDX56 Mouse Monoclonal Antibody [Clone ID: OTI1G6] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA802941 DDX56 Mouse Monoclonal Antibody [Clone ID: OTI1G6] Product data: Product Type: Primary Antibodies Clone Name: OTI1G6 Applications: WB Recommended Dilution: WB 1:2000 Reactivity: Human, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 323-547 of human DDX56 (NP_061955) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 61.4 kDa Gene Name: DEAD-box helicase 56 Database Link: NP_061955 Entrez Gene 54606 Human Q9NY93 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 DDX56 Mouse Monoclonal Antibody [Clone ID: OTI1G6] – TA802941 Background: This gene encodes a member of the DEAD box protein family. DEAD box proteins, characterized by the conserved motif Asp-Glu-Ala-Asp (DEAD), are putative RNA helicases. They are implicated in a number of cellular processes involving alteration of RNA secondary structure such as translation initiation, nuclear and mitochondrial splicing, and ribosome and spliceosome assembly. Based on their distribution patterns, some members of this family are believed to be involved in embryogenesis, spermatogenesis, and cellular growth and division. -

DDX56 (N-13): Sc-104189
SAN TA C RUZ BI OTEC HNOL OG Y, INC . DDX56 (N-13): sc-104189 BACKGROUND APPLICATIONS DEAD-box proteins, characterized by the conserved motif Asp-Glu-Ala-Asp, DDX56 (N-13) is recommended for detection of DDX56 of mouse and human are putative RNA helicases implicated in several cellular processes involving origin by Western Blotting (starting dilution 1:200, dilution range 1:100- modifications of RNA secondary structure and ribosome/spliceosome assembly. 1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein (1 ml Based on their distribution patterns, some members of this family may be of cell lysate)], immunofluorescence (starting dilution 1:50, dilution range involved in embryogenesis, spermatogenesis, and cellular growth and division. 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution range 1:30- DDX56 (DEAD box polypeptide 56), also known as DDX21 or NOH61, contains 1:3000); non cross-reactive with other DDX family members. DDX56 (N-13) a helicase core region, a leucine zipper motif in its N-terminus, two putative is also recommended for detection of DDX56 in additional species, including C-terminal nuclear localization signals and several potential phosphorylation equine, bovine and porcine. sites. DDX56 may be involved in ribosome synthesis, specifically during Suitable for use as control antibody for DDX56 siRNA (h): sc-89835, DDX56 assembly of the large 60S ribosomal subunit. siRNA (m): sc-105281, DDX56 shRNA Plasmid (h): sc-89835-SH, DDX56 shRNA Plasmid (m): sc-105281-SH, DDX56 shRNA (h) Lentiviral Particles: REFERENCES sc- 89835-V and DDX56 shRNA (m) Lentiviral Particles: sc-105281-V. -

DEAD-Box RNA Helicases in Cell Cycle Control and Clinical Therapy
cells Review DEAD-Box RNA Helicases in Cell Cycle Control and Clinical Therapy Lu Zhang 1,2 and Xiaogang Li 2,3,* 1 Department of Nephrology, Renmin Hospital of Wuhan University, Wuhan 430060, China; [email protected] 2 Department of Internal Medicine, Mayo Clinic, 200 1st Street, SW, Rochester, MN 55905, USA 3 Department of Biochemistry and Molecular Biology, Mayo Clinic, 200 1st Street, SW, Rochester, MN 55905, USA * Correspondence: [email protected]; Tel.: +1-507-266-0110 Abstract: Cell cycle is regulated through numerous signaling pathways that determine whether cells will proliferate, remain quiescent, arrest, or undergo apoptosis. Abnormal cell cycle regula- tion has been linked to many diseases. Thus, there is an urgent need to understand the diverse molecular mechanisms of how the cell cycle is controlled. RNA helicases constitute a large family of proteins with functions in all aspects of RNA metabolism, including unwinding or annealing of RNA molecules to regulate pre-mRNA, rRNA and miRNA processing, clamping protein complexes on RNA, or remodeling ribonucleoprotein complexes, to regulate gene expression. RNA helicases also regulate the activity of specific proteins through direct interaction. Abnormal expression of RNA helicases has been associated with different diseases, including cancer, neurological disorders, aging, and autosomal dominant polycystic kidney disease (ADPKD) via regulation of a diverse range of cellular processes such as cell proliferation, cell cycle arrest, and apoptosis. Recent studies showed that RNA helicases participate in the regulation of the cell cycle progression at each cell cycle phase, including G1-S transition, S phase, G2-M transition, mitosis, and cytokinesis. -

The Helicase Activity of DDX56 Is Required for Its Role in Assembly of Infectious West Nile Virus Particles
Virology 433 (2012) 226–235 Contents lists available at SciVerse ScienceDirect Virology journal homepage: www.elsevier.com/locate/yviro The helicase activity of DDX56 is required for its role in assembly of infectious West Nile virus particles Zaikun Xu a, Tom C. Hobman a,b,c,n a Department of Cell Biology, University of Alberta, 5-14 Medical Sciences Building, Edmonton, Canada T6G 2H7 b Department of Medical Microbiology and Immunology, University of Alberta, Canada T6G 2H7 c Li Ka Shing Institute of Virology, University of Alberta, Canada T6G 2H7 article info abstract Article history: Although flaviviruses encode their own helicases, evidence suggests that cellular helicases are also Received 26 April 2012 required for replication and/or assembly of these viruses. By and large, the mechanisms of action for Returned to author for revisions viral and cellular helicases are not known. Moreover, in some cases, enzymatic activity is not even 1 June 2012 required for their roles in virus biology. Recently, we showed that expression of the host nucleolar Accepted 3 August 2012 helicase DDX56 is important for infectivity of West Nile virus (WNV) particles. In the present study, we Available online 25 August 2012 demonstrate that the helicase activity of this enzyme is essential for its role in assembly of infectious Keywords: WNV virions. Over-expression of the capsid-binding region of DDX56 also reduces infectivity of WNV West Nile virus suggesting that interaction of DDX56 and capsid protein is an important step in the virion assembly Flavivirus pathway. To our knowledge, this is the first study showing that enzymatic activity of a cellular helicase Helicase is critical for infectivity of flaviviruses. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

DEAH)/RNA Helicase a Helicases Sense Microbial DNA in Human Plasmacytoid Dendritic Cells
Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells Taeil Kima, Shwetha Pazhoora, Musheng Baoa, Zhiqiang Zhanga, Shino Hanabuchia, Valeria Facchinettia, Laura Bovera, Joel Plumasb, Laurence Chaperotb, Jun Qinc, and Yong-Jun Liua,1 aDepartment of Immunology, Center for Cancer Immunology Research, University of Texas M. D. Anderson Cancer Center, Houston, TX 77030; bDepartment of Research and Development, Etablissement Français du Sang Rhône-Alpes Grenoble, 38701 La Tronche, France; and cDepartment of Biochemistry, Baylor College of Medicine, Houston, TX 77030 Edited by Ralph M. Steinman, The Rockefeller University, New York, NY, and approved July 14, 2010 (received for review May 10, 2010) Toll-like receptor 9 (TLR9) senses microbial DNA and triggers type I Microbial nucleic acids, including their genomic DNA/RNA IFN responses in plasmacytoid dendritic cells (pDCs). Previous and replicating intermediates, work as strong PAMPs (13), so studies suggest the presence of myeloid differentiation primary finding PRR-sensing pathogenic nucleic acids and investigating response gene 88 (MyD88)-dependent DNA sensors other than their signaling pathway is of general interest. Cytosolic RNA is TLR9 in pDCs. Using MS, we investigated C-phosphate-G (CpG)- recognized by RLRs, including RIG-I, melanoma differentiation- binding proteins from human pDCs, pDC-cell lines, and interferon associated gene 5 (MDA5), and laboratory of genetics and physi- regulatory factor 7 (IRF7)-expressing B-cell lines. CpG-A selectively ology 2 (LGP2). RIG-I senses 5′-triphosphate dsRNA and ssRNA bound the aspartate-glutamate-any amino acid-aspartate/histi- or short dsRNA with blunt ends. -

DDX56 Rabbit Pab
Leader in Biomolecular Solutions for Life Science DDX56 Rabbit pAb Catalog No.: A9487 Basic Information Background Catalog No. This gene encodes a member of the DEAD box protein family. DEAD box proteins, A9487 characterized by the conserved motif Asp-Glu-Ala-Asp (DEAD), are putative RNA helicases. They are implicated in a number of cellular processes involving alteration of Observed MW RNA secondary structure such as translation initiation, nuclear and mitochondrial 61kDa splicing, and ribosome and spliceosome assembly. Based on their distribution patterns, some members of this family are believed to be involved in embryogenesis, Calculated MW spermatogenesis, and cellular growth and division. The protein encoded by this gene 57kDa/61kDa shows ATPase activity in the presence of polynucleotides and associates with nucleoplasmic 65S preribosomal particles. This gene may be involved in ribosome Category synthesis, most likely during assembly of the large 60S ribosomal subunit. Multiple transcript variants encoding different isoforms have been found for this gene. Primary antibody Applications WB Cross-Reactivity Human, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 54606 Q9NY93 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 318-547 of human DDX56 (NP_061955.1). Synonyms DDX56;DDX21;DDX26;NOH61 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using DDX56 antibody (A9487) at 1:3000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. -

Integrated Analysis of DEAD-Box Helicase 56: a Potential Oncogene in Osteosarcoma
ORIGINAL RESEARCH published: 26 June 2020 doi: 10.3389/fbioe.2020.00588 Integrated Analysis of DEAD-Box Helicase 56: A Potential Oncogene in Osteosarcoma Chen Zhu 1†, Xianzuo Zhang 1*†, Nikolaos Kourkoumelis 2, Yong Shen 3,4* and Wei Huang 1 1 Division of Life Sciences and Medicine, Department of Orthopedics, The First Affiliated Hospital of USTC, University of Science and Technology of China, Hefei, China, 2 Department of Medical Physics, School of Health Sciences, University of Ioannina, Ioannina, Greece, 3 Institute on Aging and Brain Disorders, The First Affiliated Hospital of University of Science and Technology of China, Hefei, China, 4 Division of Life Sciences and Medicine, Neurodegenerative Disorder Research Center, University of Science and Technology of China, Hefei, China Background: Osteosarcoma is a solid tumor common in the musculoskeletal system. The DEAD-box helicase (DDX) families play an important role in tumor genesis and proliferation. Objective: To screen potential molecular targets in osteosarcoma and elucidate its Edited by: Bing Wang, relationship with DDX56. Anhui University of Technology, China Methods: We employed the Gene Expression Omnibus and The Cancer Genome Reviewed by: Atlas datasets for preliminary screening. DDX56 expression was measured by RT-qPCR Chun Hoi Yan, The University of Hong Kong, in three osteosarcoma cell lines. Biological roles of DDX56 were explored by Gene Hong Kong ontology, Kyoto Encyclopedia of Genes and Genomes and Ingenuity Pathway Analysis. Tao Cheng, TM Shanghai Jiao Tong University, China Cell proliferation, cycle, and apoptosis assays were performed using Lentivirus *Correspondence: knockdown technique. Xianzuo Zhang Results: It was found that DDX56 expression was regularly upregulated in osteosarcoma [email protected] Yong Shen tissue and cell lines, while DDX56 knockdown inhibited cell proliferation and promoted [email protected] cell apoptosis. -

Multiclass Analysis of Gene Expression Data the Protein Process
Critical Dynamic Processes in the Pathogenesis of Cancer Multiclass Analysis of Gene Expression Data Robert Stengel March 27, 2006 ! Background ! Small, round, blue-cell tumor example ! Analysis of colon cancer, metastases, and normal tissue presented at the Institute for Advanced Study Putative Paradigm for The Protein Process Microarray Analysis: Expression Level Infers Cell Function & Carcinogenesis ! Up-regulation in tumor cells – Causal input (tumor enhancing gene) – Defensive response (tumor suppressor gene) – Bystander effect – Tissue effect – Artifact ! Down-regulation in tumor cells – Present, but mutated (and, therefore, not detected) – Eliminated or suppressed by tumor growth – Tissue effect – Artifact Models of Gene Microarray Classification Interaction Objectives ! Inhibition and amplification are multigene effects !Class comparison ! Effects of over/underexpression depend on regulatory pathway and sign of interaction – Identify expression profiles (feature sets) for predefined classes !Class prediction – Develop mathematical function/algorithm that predicts class membership for a novel expression profile !Class discovery – Identify new classes, sub-classes, or features related to classification objectives 200 150 Tumor Typical Pairs of Colon Cancer Normal 100 Characteristics of D14657 Microarray Expression Levels 5 0 0 Classification Features -50 (Alon, Notterman, Levine et al, 1999) -20 -10 0 1 0 2 0 3 0 4 0 M94363 ! Samples not well differentiated in individual transcript clusters (overlapping) 250 200 150 Tumor Normal -
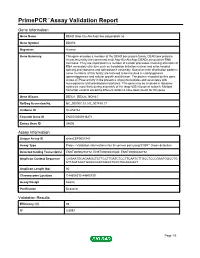
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DEAD (Asp-Glu-Ala-Asp) box polypeptide 56 Gene Symbol DDX56 Organism Human Gene Summary This gene encodes a member of the DEAD box protein family. DEAD box proteins characterized by the conserved motif Asp-Glu-Ala-Asp (DEAD) are putative RNA helicases. They are implicated in a number of cellular processes involving alteration of RNA secondary structure such as translation initiation nuclear and mitochondrial splicing and ribosome and spliceosome assembly. Based on their distribution patterns some members of this family are believed to be involved in embryogenesis spermatogenesis and cellular growth and division. The protein encoded by this gene shows ATPase activity in the presence of polynucleotides and associates with nucleoplasmic 65S preribosomal particles. This gene may be involved in ribosome synthesis most likely during assembly of the large 60S ribosomal subunit. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases DDX21, DDX26, NOH61 RefSeq Accession No. NC_000007.13, NT_007819.17 UniGene ID Hs.654762 Ensembl Gene ID ENSG00000136271 Entrez Gene ID 54606 Assay Information Unique Assay ID qHsaCEP0051943 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000258772, ENST00000431640, ENST00000448192 Amplicon Context Sequence CAGAATGCAGAAGCTCTTCCTTGATCTCCTTCAATCTTGCCTCCCGAATGGCCTG CTTAGTCACTGAGCGCATGGCATCCCTGCAGCGAT Amplicon Length (bp) 60 Chromosome Location 7:44608513-44608739 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9997 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 20.63 cDNA Tm (Celsius) 87.5 gDNA Cq 24.95 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Manipulation of Cellular Processes Via Nucleolus Hijaking in the Course of Viral Infection in Mammals
cells Review Manipulation of Cellular Processes via Nucleolus Hijaking in the Course of Viral Infection in Mammals Olga V. Iarovaia *, Elena S. Ioudinkova, Artem K. Velichko and Sergey V. Razin Institute of Gene Biology Russian Academy of Sciences, 119334 Moscow, Russia; [email protected] (E.S.I.); [email protected] (A.K.V.); [email protected] (S.V.R.) * Correspondence: [email protected]; Tel.: +7-499-135-97-87 Abstract: Due to their exceptional simplicity of organization, viruses rely on the resources, molecular mechanisms, macromolecular complexes, regulatory pathways, and functional compartments of the host cell for an effective infection process. The nucleolus plays an important role in the process of interaction between the virus and the infected cell. The interactions of viral proteins and nucleic acids with the nucleolus during the infection process are universal phenomena and have been described for almost all taxonomic groups. During infection, proteins of the nucleolus in association with viral components can be directly used for the processes of replication and transcription of viral nucleic acids and the assembly and transport of viral particles. In the course of a viral infection, the usurpation of the nucleolus functions occurs and the usurpation is accompanied by profound changes in ribosome biogenesis. Recent studies have demonstrated that the nucleolus is a multifunctional and dynamic compartment. In addition to the biogenesis of ribosomes, it is involved in regulating the cell cycle and apoptosis, responding to cellular stress, repairing DNA, and transcribing RNA polymerase II-dependent genes. A viral infection can be accompanied by targeted transport of viral Citation: Iarovaia, O.V.; Ioudinkova, proteins to the nucleolus, massive release of resident proteins of the nucleolus into the nucleoplasm E.S.; Velichko, A.K.; Razin, S.V.