Analysis of Codon Usage Patterns and Influencing Factors in Nipah Virus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

E4182 V1: Draft Final Report Vol. I
Public Disclosure Authorized Consultancy Services for Undertaking Environmental Assessment for the Rural Water Supply & Sanitation Project in Assam Public Disclosure Authorized March 2013 DRAFT FINAL REPORT VOLUME I Public Disclosure Authorized Submitted To: Chief Engineer (PHE), Assam. World Bank Project, Hengrabari,Guwahati-781036 Submitted By: IPE Global Pvt. Ltd. Public Disclosure Authorized (Formerly Infrastructure Professionals Enterprise (P) Ltd.) Address: IPE Towers, B-84, Defence Colony, Bhisham Pitamah Marg, New Delhi – 110024, India Tel: +91-11-40755920, 40755923; Fax: +91-11-24339534 Consultancy Services for Undertaking Environmental Assessment for the Rural Water Supply & Sanitation Project in Assam Draft Final Report Table of Contents Abbreviations and Acronyms ...............................................................................................................9 Executive Summary .......................................................................................................................... 11 1. INTRODUCTION ......................................................................................................................... 21 1.1 Background ....................................................................................................................... 21 1.2 Present World Bank Assisted Rural Water Supply and Sanitation Project............................... 23 1.2.1 Components............................................................................................................... 23 -

List of Acs Revenue & Election District Wise
List of Assembly Constituencies showing their Revenue & Election District wise break - up Name of the District Name of the Election Assembly Constituency Districts No. Name 1. Karimganj 1-Karimganj 1 Ratabari (SC) 2 Patharkandi 3 Karimganj North 4 Karimganj South 5 Badarpur 2. Hailakandi 2-Hailakandi 6 Hailakandi 7 Katlicherra 8 Algapur 3. Cachar 3-Silchar 9 Silchar 10 Sonai 11 Dholai (SC) 12 Udharbond 13 Lakhipur 14 Barkhola 15 Katigorah 4. Dima Hasao 4-Haflong 16 Halflong (ST) 5. Karbi Anglong 5-Bokajan 17 Bokajan (ST) 6-Diphu 18 Howraghat (ST) 19 Diphu (ST) 6. West Karbi Anglong 7-Hamren 20 Baithalangso (ST) 7. South Salmara 8-South Salmara 21 Mankachar Mankachar 22 Salmara South 8. Dhubri 9-Dhubri 23 Dhubri 24 Gauripur 25 Golakganj 26 Bilasipara West 10-Bilasipara 27 Bilasipara East 9. Kokrajhar 11-Gossaigaon 28 Gossaigaon 29 Kokrajhar West (ST) 12-Kokrajhar 30 Kokrajhar East (ST) 10. Chirang 13-Chirang 31 Sidli (ST) 14-Bijni 33 Bijni 11. Bongaigaon 15-Bogaigaon 32 Bongaigaon 16-North Salmara 34 Abhayapuri North 35 Abhayapuri South (SC) 12. Goalpara 17-Goalpara 36 Dudhnoi (ST) 37 Goalpara East 38 Goalpara West 39 Jaleswar 13. Barpeta 18-Barpeta 40 Sorbhog 43 Barpeta 44 Jania 45 Baghbor 46 Sarukhetri 47 Chenga 19-Bajali 41 Bhabanipur 42 Patacharkuchi Page 1 of 3 Name of the District Name of the Election Assembly Constituency Districts No. Name 14. Kamrup 20-Guwahati 48 Boko (SC) 49 Chaygaon 50 Palasbari 55 Hajo 21-Rangia 56 Kamalpur 57 Rangia 15. Kamrup Metro 22-Guwahati (Sadar) 51 Jalukbari 52 Dispur 53 Gauhati East 54 Gauhati West 16. -

USOF Supported Intra-District SDHQ – DHQ OFC Network for Transport of Rural/Remote Area Traffic on Bandwidth Sharing Basis
USOF supported Intra-District SDHQ – DHQ OFC network for transport of rural/remote area traffic on bandwidth sharing basis. 1. General Information: BSNL has been awarded work of setting up of Intra-District SDHQ – DHQ OFC network for transport of rural/remote area traffic on bandwidth sharing basis in Assam Licensing Area by Universal Service Obligation Funds (USOF). For setting up of the Network, an agreement has been signed between BSNL and USOF. The Agreement is valid for seven years from the effective date. As per this Agreement, SDHQ – DHQ connectivity is to be provided in all the 27 Districts covering 354 nodes consisting of District Headquarters (DHQ), Sub Divisional Headquarters (SDHQs) and few en-route locations. The Network is to be rolled out by August 2012. 2. Network Details : The network shall consist of 24 Fibre, G.652, optical fibre cable, Add- Drop Multiplexing equipment (STM-16) conforming to ITU-T Standards shall be installed at all nodes. Most of the stations shall be covered with self healing rings. 3. Sharing the bandwidth with other telecom operators : a) A maximum of 30% of the total bandwidth created under this network shall be utilised by BSNL for transporting its own traffic. The remaining bandwidth ( minimum 70%) is to be shared with other licensed telecom operators for providing services to rural / remote areas at subsidised rates. The bandwidth shall be provided on lease from SDHQ -SDHQ, having both end nodes in the same district or different districts. b) During the validity of the Agreement, BSNL shall provide bandwidth to other licensed telecom operators at a rate not more than 26.22% of the current TRAI ceiling tariffs prescribed in TRAI tariff notification of 21st April 2005. -

The Gauhati High Court
Gauhati High Court List of candidates who are provisionally allowed to appear in the preliminary examination dated 6-10-2013(Sunday) for direct recruitment to Grade-III of Assam Judicial Service SL. Roll Candidate's name Father's name Gender category(SC/ Correspondence address No. No. ST(P)/ ST(H)/NA) 1 1001 A K MEHBUB KUTUB UDDIN Male NA VILL BERENGA PART I AHMED LASKAR LASKAR PO BERENGA PS SILCHAR DIST CACHAR PIN 788005 2 1002 A M MUKHTAR AZIRUDDIN Male NA Convoy Road AHMED CHOUDHURY Near Radio Station CHOUDHURY P O Boiragimoth P S Dist Dibrugarh Assam 3 1003 A THABA CHANU A JOYBIDYA Female NA ZOO NARENGI ROAD SINGHA BYE LANE NO 5 HOUSE NO 36 PO ZOO ROAD PS GEETANAGAR PIN 781024 4 1004 AASHIKA JAIN NIRANJAN JAIN Female NA CO A K ENTERPRISE VILL AND PO BIJOYNAGAR PS PALASBARI DIST KAMRUP ASSAM 781122 5 1005 ABANINDA Dilip Gogoi Male NA Tiniali bongali gaon Namrup GOGOI P O Parbatpur Dist Dibrugarh Pin 786623 Assam 6 1006 ABDUL AMIL ABDUS SAMAD Male NA NAYAPARA WARD NO IV ABHAYAPURI TARAFDAR TARAFDAR PO ABHAYAPURI PS ABHAYAPURI DIST BONGAIGAON PIN 783384 ASSAM 7 1007 ABDUL BASITH LATE ABDUL Male NA Village and Post Office BARBHUIYA SALAM BARBHUIYA UTTAR KRISHNAPUR PART II SONAI ROAD MLA LANE SILCHAR 788006 CACHAR ASSAM 8 1008 ABDUL FARUK DEWAN ABBASH Male NA VILL RAJABAZAR ALI PO KALITAKUCHI PS HAJO DIST KAMRUP STATE ASSAM PIN 781102 9 1009 ABDUL HANNAN ABDUL MAZID Male NA VILL BANBAHAR KHAN KHAN P O KAYAKUCHI DIST BARPETA P S BARPETA STATE ASSAM PIN 781352 10 1010 ABDUL KARIM SAMSUL HOQUE Male NA CO FARMAN ALI GARIGAON VIDYANAGAR PS -

District Election Plan
GOVERNMENT OF ASSAM OFFICE OF THE DEPUTY COMMISSIONER CACHAR :: SILCHAR DISTRICT ELECTION PLAN PANCHAYAT ELECTION 2017-18 Page 1 INDEX Sl No Topic Page 1 Map of Cachar District 1 2 Introduction 1-3 3 Reservation of Z. P. Member Constituencies 4 4 Reservation of A. P. Presidents & Vice Presidents 5 Reservation of A.P. Members, G. P. Presidents & G. P. Vice 5 6-10 Presidents 6 Different Election Cell & their respective Action Plan 11-44 7 Detail of Zone & Sector-wise Polling Stations 45-136 8 Counting Plan 137 9 Important Contacts Annexure-I Page 2 DISTRICT PROFILE Cachar district is located in the southernmost part of Assam. It is bounded on the north by Barail and Jayantia hill ranges, on the south by the State of Mizoram and on the west by the districts of Hailakandi and Karimganj. The district lies between 92° 24' E and 93° 15' E longitude and 24° 22' N and 25° 8' N latitude. The total geographical area of the district is 3,786 Sq. Km. The Barak is the main river of the district and apart from that there are numerous small rivers which flow from Dima Hasao district, Manipur or Mizoram. The district is mostly made up of plains, but there are a number of hills spread across the district. Cachar receives an average annual rainfall of more than 3,000mm. The climate is Tropical wet with hot and wet summers and cool winters. The climatic condition of this district is significant for humidity in summer season and it is often intolerable. -

Tender Header Entities
GOVERNMENT OF ASSAM PUBLIC WORKS ROAD DEPARTMENT (BORDER ROADS) TENDER DOCUMENT FOR CM's Spl. Package for Barak Valley During 2013-14 TENDER DOCUMENTS (TECHNICAL) NAME OF ROAD :- Improvement of PWD Road starting from Katlicherra Town to Katlicherra Bazar(Providing GSB, WBM-3, Earth work in Cutting, Earth work in Shoulder , BM and SDBC including interlocking Concrete block Pavement) under Chief Minister's Special Package for Barak Valley during 2013-14. OFFICE OF THE CHIEF ENGINEER, PWD (BORDER ROADS & NEC WROKS) ASSAM CHANDMARI, GUWAHATI-3 1 GOVERNMENT OF ASSAM OFFICE OF THE CHIEF ENGINEER, PWD (BORDER ROADS & NEC WORKS) ASSAM, CHANDMARI, GUWAHATI-3 No. CE/BR/119/2013/35 Dated Guwahati the 25th October’ 2013 PRESS NOTICE INVITING TENDER Chief Engineer, PWD Border Roads and NEC Works, Assam, Chandmari, Guwahati-3, on behalf of Governor of Assam invites bid for the following works under CM's Spl. Package for Barak Valley during 2013-14. Details may be seen at website- http://apwd.etenders.in and also at the office of the undersigned during office hours. Sl.No. District Scope of Work Approx. Value of Work (Rs. in Crore) 1 2 3 4 1 Hailakandi Improvement of PWD Road starting from Katlicherra Town to Katlicherra Bazar(Providing GSB, WBM-3, Earth work in Cutting, Earth work in Shoulder , BM and SDBC including 1.999 interlocking Concrete block Pavement) under Chief Minister's Special Package for Barak Valley during 2013-14. 2 Hailakandi Improvement of PWD Road starting from Lala Town to Sorbanandapur (GSB, WBM-3, Earth work in Cutting, Earth work in Shoulder, BM and SDBC including Interlocking 1.999 Concrete block Pavement) under Chief Minister's Special Package for Barak Valley during 2013-14 3 Hailakandi Improvement of Road from Latakandi to Sarbanandapur via. -

S:I;Ii'****:':T R11!Iiii#;::L ;; ;,: ;;,"I.:;' I
" TlF rolrowi.q @ididates '" El1",TI*.:-ff""' **rc,"ii['"l1Tlii,?.,"J]i;..'"4 ::"::::lT'lff i:: S:i;ii'****:':t r11!iiii#;::l ;; ;,: ;;,"i.:;' i iii i""11"*:ll":; [l*"xxry.-"*i*,j,s:rj:["iiH *;l m*E#l",ik'ml*r*s':;""::""lll'xi:,i"s";jp.',:lu' m's6p;q;; mr*;::"""'"'"'"" #xtl,:#iil;*u"'ly:ttl'"'J':":r;H'ri"ir;''',*let'ei b€foG 11A.M. wLtn $s @ll inorau"..*t *.t'tt'"**n'.onfH:f:H;j'il'il;i;d; ;..eicdentorwritt€n n a'*"a *"' E"lllfiii "iii "i ' ' 'n" fi i *.r.Tf'!;i"n'#f"ril:i'!ri::h":-i T and Address of the Nahe q!!!4!C!E- Rata.pur Poad HaLl'lan6r AB6aL HELIM LAS(AR, 7, Dst. Ha'laka@L luDirl-rlHoouE BAPBHUIYA'- 5/o Mo$haraf Ali salbhuM' 3, Monachera' Vill - NischintapuFll, PO Ratanpur Road, llailaka'di Road, HalLaka'dr 5- RatanP!r RatanpurRoad, NaiakandL Ratanplr Road, Hallakandi Ratanpur Road, Hailakandi Y^ --uL Murrr CHoUDHUPY, I LT i6' r^. akr,< llddin Choldhury' L ,". ll Fo_ Prnsaur' 1i,ri-;khi6ond Pt I 1ls & Dln_ Harlakandi I E-DULRouF LAsdP' 1 ./o. abdulNoor Laskar, 1 !oad, HaLlalandi r,9o Rangau! Ratanpu ,'.lilin- Bilp.r ohumk l raLEtandi sALAM iasloR, -f H.S. School, l/O- Falar Uddin t'sl'r' I skha,,Gchar' -gDULXl ffffilXfii 1 PlN 788oo5-- | ;-BDULWAHID MPBHUTYA,- I </n-Abdu!MatnBalbhuiva' I pitanour Road HarlarandL Kanbaa i,'ili- chandlpu, prul' P'o" I AlqaPLl! Bazar. Ps- !:lahd' I ABOUL WAHID WS^, :,^- rr M,Lram AI Laskir' l RatanPUr Road, Hai!akandi U;;u;nd', raGPUr' Near PHE' sikhar' cacharTssQll-- - s/cL M.dhusudhan chanda, 1 Ratanpli Road, Hailakandi 15. -

Regions of Assam
REGIONS OF ASSAM Geographically Assam is situated in the north-eastern region of the Indian sub- continent. It covers an area of 78,523 sq. kilometres (approximate). Assam – the gateway to north-east India is a land of blue hills, valleys and rivers. Assam has lavishly bestowed upon unique natural beauty and abundant natural wealth. The natural beauty of Assam is one of the most fascinating in the country with evergreen forests, majestic rivers, rich landscape, lofty green hills, bushy grassy plains, rarest flora and fauna, beautiful islands and what not. The capital of Assam is Dispur and the state emblem is one-hoed rhino. Assam is bounded by Manipur, Nagaland and Myanmar in the east and in the rest by West Bengal in the north by Bhutan and Arunachal Pradesh and in the route by Mizoram, Tripura, Bangladesh and Meghalaya. Literacy rate in Assam has seen upward trend and is 72.19 percent as per 2011 population census. Of that, male literacy stands at 77.85 percent while female literacy is at 66.27 percent. As per details from Census 2011, Assam has population of 3.12 Crores, an increase from figure of 2.67 Crore in 2001 census. Total population of Assam as per 2011 census is 31,205,576 of which male and female are 15,939,443 and 15,266,133 respectively. In 2001, total population was 26,655,528 in which males were 13,777,037 while females were 12,878,491. The total population growth in this decade was 17.07 percent while in previous decade it was 18.85 percent. -

Status of College Library Services in Barak Valley, Assam: a Study
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Library Philosophy and Practice (e-journal) Libraries at University of Nebraska-Lincoln August 2020 Status of College Library Services in Barak Valley, Assam: A Study Sufia Khanom Mozumder USTM, Meghalaya, [email protected] P K. Barooah USTM, Meghalaya, [email protected] Mahtab Hussain USTM, Meghalaya, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/libphilprac Part of the Library and Information Science Commons Mozumder, Sufia Khanom; Barooah, P K.; and Hussain, Mahtab, "Status of College Library Services in Barak Valley, Assam: A Study" (2020). Library Philosophy and Practice (e-journal). 4011. https://digitalcommons.unl.edu/libphilprac/4011 Status of College Library Services in Barak Valley: A Study Sufia Khanom Mozumder Research Scholar, Department of Library and Information Sciences, University of Science & Technology, Baridua, Meghalaya 793101, India, E-mail: [email protected] P. K. Barooah Department of Library and Information Sciences, University of Science & Technology, Baridua, Meghalaya 793101, India, E-mail: [email protected] Mahtab Hussain Department of Library and Information Sciences, University of Science & Technology, Baridua, Meghalaya 793101, India, E-mail: [email protected] Abstract The aim of the present paper is to know the current status of various library services provided by different Under Graduate (U. G). College libraries of Barak valley, Southern Assam. The tremendous development in Information Communication Technology and their application in the various aspects of libraries have opened a new door for the libraries and users alike. In the age of Information Technology, there is a huge change in the methods and techniques of seeking the information and way of information dissemination on the other hand. -

A Study of Provincialised Degree Colleges of Barak Valley, Assam
© 2019 JETIR May 2019, Volume 6, Issue 5 www.jetir.org (ISSN-2349-5162) MANAGEMENT OF COLLEGE LIBRARY AND SERVICES: A STUDY OF PROVINCIALISED DEGREE COLLEGES OF BARAK VALLEY, ASSAM. Rofique U. Laskar Dr. Sehnara B. Choudhury Librarian, A.L.C College, Algapur. AND Librarian, G. C. College, Hailakandi, Assam, India Silchar, Cachar, Assam, India. ABSTRACT Purpose: The boom of Internet and cyber World made easy access of resources on finger tip yet library has a place of authentic resources and manual, formal reading services which can be everlasting. The services of libraries, attraction of users depends on the professional library management and service policies. Therefore the college libraries have to be well managed and served professional services to its users. This can be the only motto of modern day library services to be survived in this era of information pandemic (infodemic) world. The present study is conducted to observe the library management and library services provided in the colleges of Barak Valley which are affiliated to Assam University Silchar. There are total numbers of 24 govt. degree colleges in three districts of Assam. This study will cover whole population of 24 colleges to find out the status of library management and library services. Methodology: The present study is a survey based descriptive study with the help of structured questionnaire along with interview and personal observation to conduct this study. There are total numbers of 24 govt. degree colleges in three districts of Assam. This study will cover whole population of 24 colleges to find out the status of library management and library services. -
District Report HAILAKANDI
Baseline Survey of Minority Concentrated Districts District Report HAILAKANDI Study Commissioned by Ministry of Minority Affairs Government of India Study Conducted by Omeo Kumar Das Institute of Social Change and Development: Guwahati VIP Road, Upper Hengerabari, Guwahati 781036 1 ommissioned by the Ministry of Minority CAffairs, this Baseline Survey was planned for 90 minority concentrated districts (MCDs) identified by the Government of India across the country, and the Indian Council of Social Science Research (ICSSR), New Delhi coordinates the entire survey. Omeo Kumar Das Institute of Social Change and Development, Guwahati has been assigned to carry out the Survey for four states of the Northeast, namely Assam, Arunachal Pradesh, Meghalaya and Manipur. This report contains the results of the survey for Hailakandi district of Assam. The help and support received at various stages from the villagers, government officials and all other individuals are most gratefully acknowledged. ■ Omeo Kumar Das Institute of Social Change and Development is an autonomous research institute of the ICSSR, New Delhi and Government of Assam. 2 CONTENTS BACKGROUND....................................................................................................................................8 METHODOLOGY.................................................................................................................................9 TOOLS USED ......................................................................................................................................10 -
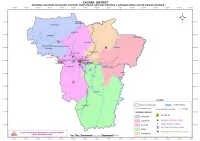
District: Cachar Information on Railway Stations, Ferryghats, Astc Bus Stations & Grounds Which Can Be Used As Helipads
DISTRICT: CACHAR INFORMATION ON RAILWAY STATIONS, FERRYGHATS, ASTC BUS STATIONS & GROUNDS WHICH CAN BE USED AS HELIPADS RAILWAY STATIONS SL. NAME REVENUE GAON PANCHAYAT VILLAGE NAME NO CIRCLE SL. NO NAME REVENUE CIRCLE GAON PANCHAYAT VILLAGE NAME 14 JATINGA FERRY SERVICE SILCHAR KUMARPARA NIZ-JOYNAGAR NIZ-JOYNAGAR 0 BIHARA RAILWAY STATION KATIGORA GORERVITAR BEHARA PT I 15 GANDHIGHAT FERRY SERVICE SILCHAR DUDPATIL DUDHPATIL PT VI 1 PANCHGRAM RAILWAY STATION ALGAPUR PANCHGRAM PANCHGRAM 2 ARUNACHAL RAILWAY STATION SILCHAR KUMARPARA NIZ-JOYNAGAR BALIGHAT ASTC BUS STATIONS 3 SILCHAR TARAPUR RAILWAY STATION SILCHAR SILCHAR TOWN WARD 27 SL. NO NAME REVENUE CIRCLE GAON PANCHAYAT VILLAGE NAME 4 SALCHAPRA RAILWAY STATION SILCHAR SALCHAPRA SALCHAPRA PT I 0 ISBT SILCHAR SILCHAR RAMNAGAR TARAPUR TARAPUR PT V 1 IS TRUCK TERMINUS SILCHAR RAMNAGAR TARAPUR TARAPUR PT V FERRY GHATS 2 ASTC STAND SILCHAR SILCHAR TOWN WARD 17 SL. NAME REVENUE GAON PANCHAYAT VILLAGE NAME NO CIRCLE 3 KUMBHIRGRAM AIRPORT UDARBOND SALGANGA KUMBHIRGRAM T.E. 0 CHOTTO- DUDHPATIL FERRY SERVICE SILCHAR MASUGHAT DUDHPATIL PT V 4 DEVDOOT STAND,SILCHAR SILCHAR SILCHAR TOWN WARD 24 1 MASIMPUR FERRY SERVICE SILCHAR BADARPUR MASIMPUR MASIMPUR PT I 5 CTVOA STAND SILCHAR SILCHAR TOWN WARD 24 (MAOHIMPUR) 2 BORO-DUDHPATIL FERRY SERVICE SILCHAR CHOTO DUDPATIL DUDHPATIL PT I 3 GANGAPUR FERRY SERVICE LAKHIPUR BADRIPAR GANGAPUR 4 SILGHAT FERRY SERVICE SONAI GOBINDAPUR ALGAPUR GOBINDAPUR PT III GROUNDS WHICH CAN BE USED AS HELIPADS 5 ANNAPURNA FERRY SERVICE SILCHAR SILCHAR TOWN WARD 24 SL. NAME