Autoimmune Liver Disease: Overlap and Outliers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Viral Hepatitis Testing Effective Date: January 1, 2012
Viral Hepatitis Testing Effective Date: January 1, 2012 Scope This guideline provides guidance for the use of laboratory tests to diagnose acute and chronic viral hepatitis in adults (> 19 years) in the primary care setting. General Considerations for Ordering Laboratory Tests Prior to ordering tests for hepatitis, consider the patient’s history, age, risk factors (see below), hepatitis vaccination status, and any available previous hepatitis test results. Risk Factors for Viral Hepatitis include: • Substance use (includes sharing drug snorting, smoking or injection equipment) • High-risk sexual activity or sexual partner with viral hepatitis • Travel to or from high-risk hepatitis endemic areas or exposure during a local outbreak • Immigration from hepatitis B and/or C endemic countries • Household contact with an infected person especially if personal items (e.g., razors, toothbrushes, nail clippers) are shared • Recipient of unscreened blood products* • Needle-stick injury or other occupational exposure (e.g., healthcare workers) • Children born to mothers with chronic hepatitis B or C infection • Attendance at daycare • Contaminated food or water (hepatitis A only) • Tattoos and body piercing • History of incarceration • HIV or other sexually transmitted infection • Hemodialysis *screening of donated blood products for hepatitis C (anti-HCV) began in 1990 in Canada.1 Types of Viral Hepatitis Hepatitis A: causes acute but not chronic hepatitis Hepatitis B: causes acute and chronic hepatitis Hepatitis C: causes chronic hepatitis but rarely manifests as acute hepatitis Hepatitis D: rare and only occurs in patients infected with hepatitis B Hepatitis E: clinically similar to hepatitis A, mostly restricted to endemic areas and occasionally causes chronic infection in immunosuppressed people Others: e.g. -

Autoimmune Hepatitis
Page 1 of 5 Autoimmune Hepatitis Autoimmune hepatitis is an uncommon cause of chronic hepatitis (persistent liver inflammation). The cause is not known. If left untreated, the inflammation causes cirrhosis (scarring of the liver). However, with treatment, the outlook for people with this condition is very good. Treatment is usually with steroids and other medicines which suppress inflammation. What does the liver do? The liver is in the upper right part of the abdomen. It has many functions which include: Storing glycogen (fuel for the body) which is made from sugars. When required, glycogen is broken down into glucose which is released into the bloodstream. Helping to process fats and proteins from digested food. Making proteins that are essential for blood to clot (clotting factors). Processing many medicines which you may take. Helping to remove or process alcohol, poisons and toxins from the body. Making bile which passes from the liver to the gut down the bile duct. Bile breaks down the fats in food so that they can be absorbed from the bowel. What is autoimmune hepatitis? Hepatitis means inflammation of the liver. There are many causes of hepatitis. For example, alcohol excess and infections with various viruses are the common causes of hepatitis. Autoimmune hepatitis is an uncommon cause of chronic hepatitis. Chronic means that the inflammation is persistent or long-term. The chronic inflammation gradually damages the liver cells, which can result in serious problems. What causes autoimmune hepatitis? Page 2 of 5 The cause is not clear. It is thought to be an autoimmune disease. Our immune system normally defends us against infection from bacteria, viruses and other germs. -

Prevention & Control of Viral Hepatitis Infection
Prevention & Control of Viral Hepatitis Infection: A Strategy for Global Action © World Health Organization 2011. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by WHO to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either express or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. Table of contents Disease burden 02 What is viral hepatitis? 05 Prevention & control: a tailored approach 06 Global Achievements 08 Remaining challenges 10 World Health Assembly: a mandate for comprehensive prevention & control 13 WHO goals and strategy -

EASL Clinical Practice Guidelines: Autoimmune Hepatitisq
Clinical Practice Guidelines EASL Clinical Practice Guidelines: Autoimmune hepatitisq ⇑ European Association for the Study of the Liver striking progress, and now patients in specialised centres have an Introduction excellent prognosis, both in respect to survival and to quality of life. Autoimmune hepatitis (AIH) was the first liver disease for which The aim of the present Clinical Practice Guideline (CPG) is to an effective therapeutic intervention, corticosteroid treatment, provide guidance to hepatologists and general physicians in the was convincingly demonstrated in controlled clinical trials. diagnosis and treatment of AIH in order to improve care for However, 50 years later AIH still remains a major diagnostic affected patients. In view of the limited data from large con- and therapeutic challenge. There are two major reasons for this trolled studies and trials, many recommendations are based on apparent contradiction: Firstly, AIH is a relatively rare disease. expert consensus. This is to some extent a limitation of this Secondly, AIH is a very heterogeneous disease. EASL-CPG, but at the same time it is its special strength: consen- Like other rare diseases, clinical studies are hampered by the sus in this guideline is based on intensive discussions of experts limited number of patients that can be included in trials. Possibly from large treatment centres. The core consensus group has and more importantly, the interest of the pharmaceutical indus- experience of over one thousand AIH patients managed person- try to develop effective specific therapies for rare diseases is lim- ally, and the recommendations have been reviewed by both the ited due to the very restricted market for such products. -

AASLD PRACTICE GUIDELINES Diagnosis and Management of Autoimmune Hepatitis
AASLD PRACTICE GUIDELINES Diagnosis and Management of Autoimmune Hepatitis Michael P. Manns,1 Albert J. Czaja,2 James D. Gorham,3 Edward L. Krawitt,4 Giorgina Mieli-Vergani,5 Diego Vergani,6 and John M. Vierling7 This guideline has been approved by the American ment on Guidelines;3 and (4) the experience of the Association for the Study of Liver Diseases (AASLD) authors in the specified topic. and represents the position of the Association. These recommendations, intended for use by physi- cians, suggest preferred approaches to the diagnostic, 1. Preamble therapeutic and preventive aspects of care. They are intended to be flexible, in contrast to standards of Clinical practice guidelines are defined as ‘‘systemati- care, which are inflexible policies to be followed in ev- cally developed statements to assist practitioner and ery case. Specific recommendations are based on rele- patient decisions about appropriate heath care for spe- vant published information. To more fully characterize 1 cific clinical circumstances.’’ These guidelines on the quality of evidence supporting the recommenda- autoimmune hepatitis provide a data-supported tions, the Practice Guidelines Committee of the approach to the diagnosis and management of this dis- AASLD requires a class (reflecting benefit versus risk) ease. They are based on the following: (1) formal and level (assessing strength or certainty) of evidence review and analysis of the recently-published world lit- to be assigned and reported with each recommenda- erature on the topic [Medline search]; (2) American tion.4 The grading system applied to the recommenda- College of Physicians Manual for Assessing Health tions has been adapted from the American College of 2 Practices and Designing Practice Guidelines; (3) Cardiology and the American Heart Association Prac- guideline policies, including the AASLD Policy on the tice Guidelines, and it is given below (Table 1). -

Chronic Viral Hepatitis in a Cohort of Inflammatory Bowel Disease
pathogens Article Chronic Viral Hepatitis in a Cohort of Inflammatory Bowel Disease Patients from Southern Italy: A Case-Control Study Giuseppe Losurdo 1,2 , Andrea Iannone 1, Antonella Contaldo 1, Michele Barone 1 , Enzo Ierardi 1 , Alfredo Di Leo 1,* and Mariabeatrice Principi 1 1 Section of Gastroenterology, Department of Emergency and Organ Transplantation, University “Aldo Moro” of Bari, 70124 Bari, Italy; [email protected] (G.L.); [email protected] (A.I.); [email protected] (A.C.); [email protected] (M.B.); [email protected] (E.I.); [email protected] (M.P.) 2 Ph.D. Course in Organs and Tissues Transplantation and Cellular Therapies, Department of Emergency and Organ Transplantation, University “Aldo Moro” of Bari, 70124 Bari, Italy * Correspondence: [email protected]; Tel.: +39-080-559-2925 Received: 14 September 2020; Accepted: 21 October 2020; Published: 23 October 2020 Abstract: We performed an epidemiologic study to assess the prevalence of chronic viral hepatitis in inflammatory bowel disease (IBD) and to detect their possible relationships. Methods: It was a single centre cohort cross-sectional study, during October 2016 and October 2017. Consecutive IBD adult patients and a control group of non-IBD subjects were recruited. All patients underwent laboratory investigations to detect chronic hepatitis B (HBV) and C (HCV) infection. Parameters of liver function, elastography and IBD features were collected. Univariate analysis was performed by Student’s t or chi-square test. Multivariate analysis was performed by binomial logistic regression and odds ratios (ORs) were calculated. We enrolled 807 IBD patients and 189 controls. Thirty-five (4.3%) had chronic viral hepatitis: 28 HCV (3.4%, versus 5.3% in controls, p = 0.24) and 7 HBV (0.9% versus 0.5% in controls, p = 0.64). -

Primary Biliary Cholangitis: a Brief Overview Justin S
REVIEW Primary Biliary Cholangitis: A Brief Overview Justin S. Louie,* Sirisha Grandhe,* Karen Matsukuma,† and Christopher L. Bowlus* Primary biliary cholangitis (PBC), previously referred to supported by the higher concordance of PBC in monozy- as primary biliary cirrhosis, is the most common chronic gotic compared with dizygotic twins.4 In addition, certain cholestatic autoimmune disease affecting adults in the human leukocyte antigen haplotypes have been associ- United States.1 It is characterized by a hallmark serologic ated with PBC, as well as variants at loci along the inter- signature, antimitochondrial antibody (AMA), and specific leukin-12 (IL-12) immunoregulatory pathway (IL-12A and bile duct pathology with progressive intrahepatic duct de- IL-12RB2 loci).5 struction leading to cholestasis. PBC is potentially fatal and can have both intrahepatic and extrahepatic complications. PATHOGENESIS EPIDEMIOLOGY The primary disease mechanism in PBC is thought to be T cell lymphocyte–mediated injury against intralobu- PBC affects all races and ethnicities; however, it is best lar biliary epithelial cells. This causes progressive destruc- studied in the Caucasian population. The condition pre- tion and eventual disappearance of the intralobular bile dominantly affects women older than 40 years, with a ducts. Molecular mimicry has been proposed as the ini- female/male ratio of 9:1.2 Although the incidence of PBC tiating event in the loss of tolerance primarily to mito- appears to be stable, the overall prevalence of the disease chondrial pyruvate dehydrogenase complex, E2, during is increasing.3 An individual’s genetic susceptibility, epige- which exogenous antigens evoke an immune response netic factors, and certain environmental triggers seem to that recognizes an endogenous (self) antigen inciting an play important roles. -
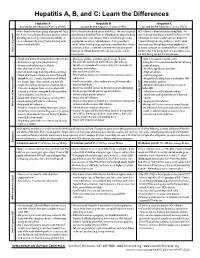
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Active Peptic Ulcer Disease in Patients with Hepatitis C Virus-Related Cirrhosis: the Role of Helicobacter Pylori Infection and Portal Hypertensive Gastropathy
dore.qxd 7/19/2004 11:24 AM Page 521 View metadata, citation and similar papers at core.ac.uk ORIGINAL ARTICLE brought to you by CORE provided by Crossref Active peptic ulcer disease in patients with hepatitis C virus-related cirrhosis: The role of Helicobacter pylori infection and portal hypertensive gastropathy Maria Pina Dore MD PhD, Daniela Mura MD, Stefania Deledda MD, Emmanouil Maragkoudakis MD, Antonella Pironti MD, Giuseppe Realdi MD MP Dore, D Mura, S Deledda, E Maragkoudakis, Ulcère gastroduodénal évolutif chez les A Pironti, G Realdi. Active peptic ulcer disease in patients patients atteints de cirrhose liée au HCV : Le with hepatitis C virus-related cirrhosis: The role of Helicobacter pylori infection and portal hypertensive rôle de l’infection à Helicobacter pylori et de la gastropathy. Can J Gastroenterol 2004;18(8):521-524. gastropathie liée à l’hypertension portale BACKGROUND & AIM: The relationship between Helicobacter HISTORIQUE ET BUT : Le lien entre l’infection à Helicobacter pylori pylori infection and peptic ulcer disease in cirrhosis remains contro- et l’ulcère gastroduodénal dans la cirrhose reste controversé. Le but de la versial. The purpose of the present study was to investigate the role of présente étude est de vérifier le rôle de l’infection à H. pylori et de la gas- H pylori infection and portal hypertension gastropathy in the preva- tropathie liée à l’hypertension portale dans la prévalence de l’ulcère gas- lence of active peptic ulcer among dyspeptic patients with compen- troduodénal évolutif chez les patients dyspeptiques souffrant d’une sated hepatitis C virus (HCV)-related cirrhosis. -

Understanding Human Astrovirus from Pathogenesis to Treatment
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 6-2020 Understanding Human Astrovirus from Pathogenesis to Treatment Virginia Hargest University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Diseases Commons, Medical Sciences Commons, and the Viruses Commons Recommended Citation Hargest, Virginia (0000-0003-3883-1232), "Understanding Human Astrovirus from Pathogenesis to Treatment" (2020). Theses and Dissertations (ETD). Paper 523. http://dx.doi.org/10.21007/ etd.cghs.2020.0507. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Understanding Human Astrovirus from Pathogenesis to Treatment Abstract While human astroviruses (HAstV) were discovered nearly 45 years ago, these small positive-sense RNA viruses remain critically understudied. These studies provide fundamental new research on astrovirus pathogenesis and disruption of the gut epithelium by induction of epithelial-mesenchymal transition (EMT) following astrovirus infection. Here we characterize HAstV-induced EMT as an upregulation of SNAI1 and VIM with a down regulation of CDH1 and OCLN, loss of cell-cell junctions most notably at 18 hours post-infection (hpi), and loss of cellular polarity by 24 hpi. While active transforming growth factor- (TGF-) increases during HAstV infection, inhibition of TGF- signaling does not hinder EMT induction. However, HAstV-induced EMT does require active viral replication. -

Hepatitis C 2005 Clinical Guidelines Summary of The: New York State Department of Health Clinical Guidelines for the Medical Management of Hepatitis C
Hepatitis C 2005 Clinical Guidelines Summary of the: New York State Department of Health Clinical Guidelines for the Medical Management of Hepatitis C Inside: Key Features of Viral Hepatitis A,B and C 1 Natural Course of HCV Infection 2 Persons at Risk for HCV Infection 3 Sources of HCV Infection 4 Counseling Prior To Testing 5 Screening for HCV Algorithm 6 Laboratory Testing for HCV 7 Interpretation of HCV Test Results 8 Post Exposure Screening for HCV 9 Counseling After Testing 10 Treating HCV Patients 11 Medical Management of HCV Positive Patients 12 References and Internet Resources 13 Hepatitis C Virus The Hidden Epidemic The Burden of HCV • 3 million Americans are chronically infected with the • 8-10,000 deaths a year are caused by HCV. Hepatitis C virus (HCV). • HCV is the leading cause for liver transplants and • 342,000 New Yorkers are estimated to be infected chronic liver disease. with HCV. • HCV deaths will increase four-fold to 38,000, by • A majority of the people infected with HCV do not the year 2010. know they have it. • Years of life lost to Hepatitis C (2001-2019) • Thousands of people go undetected each year—due 3.1 million years to inadequate risk assessment, under-screening and confusion about the use of diagnostic tests. • Cost of premature disability and death (2010-2109) $75.5 billion Hepatitis C Virus • Direct medical costs in absentee losses due to Hepatitis C $750 million/ year • HCV is a blood-borne disease transmitted by • Total medical expenditures for persons with HCV blood-to-blood contact. -

Hepatitis C Virus Infection and Human Pancreatic ß-Cell Dysfunction
Pathophysiology/Complications BRIEF REPORT Hepatitis C Virus Infection and Human Pancreatic -Cell Dysfunction 1 1 MATILDE MASINI, MD SILVIA DEL GUERRA, PHD showing the characteristic insulin gran- 2 1 DANIELA CAMPANI, MD MARCO BUGLIANI, PHD ules and normally preserved mitochon- 3 1 UGO BOGGI, MD SCILLA TORRI, PHD  2 1 dria. In -cells from HCV-positive MICHELE MENICAGLI, MD STEFANO DEL PRATO, MD 1 3 pancreases, the presence of virus-like par- NICOLA FUNEL, MD FRANCO MOSCA, MD 1 4 ticles was observed, mainly close to the MARIA POLLERA, MD FRANCO FILIPPONI, MD 1 1 membranes of Golgi apparatus, which, in ROBERTO LUPI, PHD PIERO MARCHETTI, MD, PHD turn, appeared hyperplastic and dilated (Fig. 1D). The mitochondria appeared round-shaped with dispersed matrix and fragmented cristae (Fig. 1D). Additional Ϯ 2 any patients with chronic hepati- and 2 women, BMI 25.8 1.6 kg/m ) -cell changes were observed at the Ϯ tis C virus (HCV) develop type 2 and 10 HCV-negative (age 67 9 years, level of rough endoplasmic reticulum, Ϯ diabetes (1). This prevalence is 6 men and 4 women, BMI 26.8 2.0 which showed long and dilated tubular M 2 much higher than that observed in the kg/m ) donors were harvested and stud- membranes, with numerous electron- general population and in patients with ied with the approval of our local ethics dense ribosomes bound to the latter (not other chronic liver diseases such as hepa- committee. Histological studies were shown). These morphological changes titis B virus, alcoholic liver disease, and performed by immunohistochemistry were accompanied by reduced in vitro primary biliary cirrhosis.