Cell-Specific Roles for CASK in the Pathology of Optic Nerve Hypoplasia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Update on Progress and the Changing Epidemiology of Causes of Childhood Blindness Worldwide
Major Articles An update on progress and the changing epidemiology of causes of childhood blindness worldwide Lingkun Kong, MD, PhD,a Melinda Fry, MPH,b Mohannad Al-Samarraie, MD,a Clare Gilbert, MD, FRCOphth,c and Paul G. Steinkuller, MDa PURPOSE To summarize the available data on pediatric blinding disease worldwide and to present current information on childhood blindness in the United States. METHODS A systematic search of world literature published since 1999 was conducted. Data also were solicited from each state school for the blind in the United States. RESULTS In developing countries, 7% to 31% of childhood blindness and visual impairment is avoidable, 10% to 58% is treatable, and 3% to 28% is preventable. Corneal opacification is the leading cause of blindness in Africa, but the rate has decreased significantly from 56% in 1999 to 28% in 2012. There is no national registry of the blind in the United States, and most schools for the blind do not maintain data regarding the cause of blindness in their students. From those schools that do have such information, the top three causes are cortical visual impairment, optic nerve hypoplasia, and retinopathy of prematurity, which have not changed in past 10 years. CONCLUSIONS There are marked regional differences in the causes of blindness in children, apparently based on socioeconomic factors that limit prevention and treatment schemes. In the United States, the 3 leading causes of childhood blindness appear to be cortical visual impairment, optic nerve hypoplasia, and retinopathy of prematurity; a national registry of the blind would allow accumulation of more complete and reliable data for accurate determination of the prevalence of each. -

Review Article the Status of Childhood Blindness And
[Downloaded free from http://www.meajo.org on Wednesday, January 07, 2015, IP: 41.235.88.16] || Click here to download free Android application for this journal Review Article The Status of Childhood Blindness and Functional Low Vision in the Eastern Mediterranean Region in 2012 Rajiv Khandekar, H. Kishore1, Rabiu M. Mansu2, Haroon Awan3 ABSTRACT Access this article online Website: Childhood blindness and visual impairment (CBVI) are major disabilities that compromise www.meajo.org the normal development of children. Health resources and practices to prevent CBVI are DOI: suboptimal in most countries in the Eastern Mediterranean Region (EMR). We reviewed the 10.4103/0974-9233.142273 magnitude and the etiologies of childhood visual disabilities based on the estimates using Quick Response Code: socioeconomic proxy indicators such as gross domestic product (GDP) per capita and <5‑year mortality rates. The result of these findings will facilitate novel concepts in addressing and developing services to effectively reduce CBVI in this region. The current study determined the rates of bilateral blindness (defined as Best corrected visual acuity(BCVA)) less than 3/60 in the better eye or a visual field of 10° surrounding central fixation) and functional low vision (FLV) (visual impairment for which no treatment or refractive correction can improve the vision up to >6/18 in a better eye) in children <15 years old. We used the 2011 population projections, <5‑year mortality rates and GDP per capita of 23 countries (collectively grouped as EMR). Based on the GDP, we divided the countries into three groups; high, middle‑ and low‑income nations. -

A Survey of Visual Impairment in Children Attending the Royal Blind
Eye (2002) 16, 557–561 2002 Nature Publishing Group All rights reserved 0950-222X/02 $25.00 www.nature.com/eye J Alagaratnam, TK Sharma, CS Lim and CLINICAL STUDY A survey of visual BW Fleck impairment in children attending the Royal Blind School, Edinburgh using the WHO childhood visual impairment database Abstract Introduction Purpose To assess the aetiology and The prevalence and major causes of childhood changing patterns of childhood blindness in blindness vary between countries and over one school for the blind in the UK and to time. The WHO Prevention of Blindness assess the use of the World Health Program with the International Centre for Eye Organisation Prevention of Blindness Health has developed a standard methodology (WHO/PBL) methodology and reporting form and reporting form to record the causes of in a developed country. visual impairment in children.1 The Methods One hundred and seven children methodology has been used in developing in one school for the blind and visually and middle income countries.1–3 impaired in Edinburgh were examined using This study was undertaken in order to the WHO/PBL childhood blindness assess this method in one school for blind assessment form. children in the UK. There have been two Results Of the 107 children examined, 87 previous surveys of visual impairment in The (81%) were blind or severely visually Royal Blind School, Edinburgh.4,5 These impaired (corrected visual acuity of Ͻ6/60 studies and studies from other developed (20/200) in the better eye). Perinatal related countries6 were presented in a non- blindness (40%), hereditary disease (26%) standardised way, which makes it difficult to Department of and developmental factors (26%) formed the monitor changing patterns of childhood Ophthalmology three largest aetiological categories. -

JMSCR Vol||08||Issue||02||Page 208-210||February 2020
JMSCR Vol||08||Issue||02||Page 208-210||February 2020 http://jmscr.igmpublication.org/home/ ISSN (e)-2347-176x ISSN (p) 2455-0450 DOI: https://dx.doi.org/10.18535/jmscr/v8i2.40 Keratomalacia - A Case Report Authors Deepthi Pullepu1*, Deepthi Janga2, V Murali Krishna3 *Corresponding Author Deepthi Pullepu Department of Ophthalmology, Rangaraya Medical College, Kakinada, Andhra Pradesh, India Abstract Xerophthalmia refers to an ocular condition of destructive dryness of conjunctiva and cornea caused due to severe vitamin A deficiency. Usually affects infants, children and women of reproductive age group. It is estimated to affect about one-third of children under the age of five around the world. Keratomalacia means dry and cloudy cornea which melts and perforates, caused due to severe vitamin A deficiency. Here we describe a case of 21 year old female with neurofibromatosis type 1 presenting with keratomalacia. Keywords: Vitamin A deficiency, Xerophthalmia, Keratomalacia. Introduction blindness is a significant contributor Vitamin A is essential for the functioning of the In India, it is estimated that there are immune system, healthy growth and development approximately 6.8 million people who have vision of body and is usually acquired through diet. less than 6/60 in at least one eye due to corneal Globally, 190 million children under five years of diseases; of these, about a million have bilateral age are affected by vitamin A deficiency involvement. They suffer from an increased risk of visual Corneal blindness resulting due to this disease can impairment, illness and death from childhood be completely prevented by institution of effective infections such as measles and those causing preventive or prophylactic measures at the diarrhoea community level. -

Retinitis Pigmentosa: a Brief Review of the Genetic and Clinical Aspects
Retinitis Pigmentosa: A Brief Review of the Genetic and Clinical Aspects of the Disease Itia Dowdell Science and Technology Honors Program, University of Alabama at Birmingham, Birmingham, AL, USA School of Health Professions Honors Program, University of Alabama at Birmingham, Birmingham, AL, USA Department of Clinical and Diagnostic Sciences, University of Alabama at Birmingham, Birmingham, AL, USA Abstract Retinitis Pigmentosa (RP) is a heterogeneous set of inherited retinal diseases that affects 1 in 3,000–7,000 people worldwide. Typical onset is from 10–30 years old and most forms are progressive, often leading to blindness. Defects in more than 200 genes have been identified that cause RP. The disease is characterized as a progressive rod-cone dystrophy that presents with night blindness, loss of peripheral vision, waxy pallor of the optic disc, pigmentary changes, and a reduced visual field. There are different modes of transmission of RP: autosomal dominant (ADRP), autosomal recessive (arRP), X-linked (XLRP) and mitochondrial. The genetics behind the different forms of RP and the degree of severity vary, although some overlap, thus contributing to the difficulty of differential diagnosis. RP can manifest either as a non-syndromic disease, or as part of a syndrome, such as in Usher’s syndrome (hearing and vision loss) and BardetBiedl syndrome (a ciliopathy). The purpose of this review is to summarize the major genetic and molecular findings, as well as the diseases, associated with RP. Due to space limitations, this review is not fully comprehensive. Keywords: Retinitis pigmentosa, non-syndromic retinitis pigmentosa, rod-cone dystrophy, rhodopsin Introduction Retinitis pigmentosa (RP) is a heterogeneous set of inherited retinal diseases. -

Update/Le Point
Update/Le point Childhood blindness: a new form for recording causes of visual loss in children* C. Gilbert,1 A. Foster,' A.-D. Negrel,2 & B. Thylefors2 The new standardized form for recording the causes of visual loss in children is accompanied by coding instructions and by a database for statistical analysis. The aim is to record the causes of child- hood visual loss, with an emphasis on preventable and treatable causes, so that appropriate control measures can be planned. With this standardized methodology, it will be possible to monitor the changing patterns of childhood blindness over a period of time in response to changes in health care services, specific interventions, and socioeconomic development. Introduction countries to approximately 1.0-1.5 per 1000 children in very poor communities (3). The available informa- A standard classification for reporting the categories tion on the causes of visual loss in childhood shows of visual loss was developed in 1975 and has been marked regional variations; corneal scarring second- widely used to facilitate the comparison of data from ary to vitamin A deficiency, measles infection or different locations (1). The World Health Organiza- ophthalmia neonatorum is the main cause of child- tion's Programme for the Prevention of Blindness hood blindness in Africa and Asia, while retinal dis- (WHO/PBL) has also developed a standard protocol ease and lesions of the central nervous system are for reporting the causes of visual loss.a more important causes in Europe and North America (4). In May 1990 a global meeting on the Prevention Difficulties in comparing the causes of visual of Childhood Blindness, convened by WHO at the loss in children are due to lack of a standardized Intemational Centre for Eye Health (ICEH) in reporting system which takes into account both ana- London, recommended that further information tomical and etiological classifications. -

Abstract: a 19 Year Old Male Was Diagnosed with Vitamin a Deficiency
Robert Adam Young Abstract: A 19 year old male was diagnosed with vitamin A deficiency (VAD). Clinical examination shows conjunctival changes with central and marginal corneal ulcers. Patient history and lab testing were used to confirm the diagnosis. I. Case History 19 year old Hispanic Male Presents with chief complaint of progressive blur at distance and near in both eyes, foreign body sensation, ocular pain, photophobia, and epiphora; signs/symptoms are worse in the morning upon wakening. Started 6 months to 1 year ago, and has progressively gotten worse. Patient reports that the right eye is worse than the left eye. Ocular history of Ocular Rosacea Medical history of Hypoaldosteronism, Pernicious Anemia, and Type 2 Polyglandular Autoimmune Syndrome Last eye examination was two weeks ago at a medical center Presenting topical/systemic medications - Tobramycin TID OU, Prednisolone Acetate QD OU (has used for two weeks); Fludrocortisone (used long-term per PCP) Other pertinent info includes reports that patient cannot gain weight, although he has a regular appetite. Patient presents looking very slim, malnourished, and undersized for his age. II. Pertinent findings Entering unaided acuities are 20/400 OD with no improvement with pinhole; 20/50 OS that improves to 20/25 with pinhole. Pupil testing shows (-)APD; Pupils are 6mm in dim light, and constrict to 4mm in bright light. Ocular motilities are full and smooth with no reports of diplopia or pain. Tonometry was performed with tonopen and revealed intraocular pressures of 12 mmHg OD and 11 mmHG OS. Slit lamp examination shows 2+ conjunctival injection with trace-mild bitot spots both nasal and temporal, OU. -
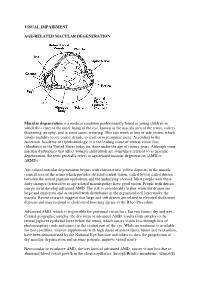
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

Profile of Pediatric Ocular Morbidity in the In-Patient Department of an Apex Public Hospital
Open Access Austin Journal of Clinical Ophthalmology A Austin Full Text Article Publishing Group Research Article Profile of Pediatric Ocular Morbidity in the in-Patient Department of an Apex Public Hospital Hossain Ava1,2*, Hussain AHME2, Ferdausi N2 and Akand AR2 Abstract 1Green Life Medical College, Dhaka,Bangladesh. Background: This study aimed at determining the causes of pediatric ocular 2National Institute of Ophthalmology & Hospital, Dhaka morbidity in the in-patient department of an apex public hospital in Bangladesh -1207, Bangladesh. where dedicated pediatric ophthalmology services have been established with *Corresponding author: Ava Hossain, Principal cum the collaboration of Government and International NGOs. Professor of Ophthalmology Green Life Medical College, Patients and Methods: This was a descriptive study using cross sectional 32 Green Road, Dhaka-1205, Bangladesh data. All patients admitted in pediatric ophthalmology department during the Received: July 25, 2014; Accepted: August 20, 2014; period of January to June, 2014 were enrolled in this study. Age at the time of Published: August 22, 2014 noticing the symptom first, time of reporting to hospital, current age and gender of the children were recorded. The clinical diagnosis was grouped according to disease category. Results were analyzed using SPSS version 17. Results: A total of 676 children were admitted for treatment of 902 eyes in pediatric ophthalmology department during the study period. There were 372 (55%) boys and 304 (45%) girls, resulting boys to girl’s ratio of 1.2:1. In this study, the most common type of disease was childhood cataract, constituting 48.28% of the admitted patients, followed by ocular injury (21.76%), ptosis (5.16%), strabismus (4.96%) and retinoblastoma (3.85%). -

Prevention of Childhood Blindness Teaching Set
INTERNATIONAL CENTRE FOR EYE HEALTH Prevention of Childhood Blindness Teaching Set © 1998, updated 2007, International Centre for Eye Health, London School of Hygiene & Tropical Medicine, Keppel Street, London, WC1E 7HT, UK. Web sites: www.iceh.org.uk and www.jceh.co.uk. Supported by CBM International, HelpAge International, Sight Savers International, Task Force Sight and Life. Table of Contents 1. Childhood Blindness Worldwide 2 2. Causes of Childhood Blindness 3 3. Onset of Blindness 4 4. Examination for Eye Disease in Children 5 5. Vitamin A Deficiency and the Eye 6 6. Symptoms and Signs of Xerophthalmia 8 7. Treatment of Xerophthalmia 10 8. Prevention of Xerophthalmia 12 9. Measles and Corneal Ulceration 14 10. Prevention and Treatment of Measles 15 11. Herpes Simplex Virus 16 12. Harmful Traditional Eye Medicines 17 13. Newborn Conjunctivitis 19 14. Treatment of Newborn Conjunctivitis 21 15. Corneal Ulceration 22 16. Corneal Scarring 25 17. Congenital Cataract 26 18. Causes and Investigation of Congenital Cataract 27 19. Surgery for Congenital Cataract 29 20. Congenital Glaucoma 31 21. Retinoblastoma 33 22. Retinopathy of Prematurity 35 23. Eye Injuries 37 24. Summary 39 Acknowledgments 41 1 1. Childhood Blindness Worldwide 600,000 39% 500,000 34% 400,000 21% 300,000 200,000 Numbers of blind Numberschildren of blind 6% 100,000 0 Rich Middle Poor Very poor Standards of living and health care services How many children in the world are blind? The exact number of children blind in the world is not known but it is estimated that the figure is approximately 1.4 million, with up to 500,000 new cases every year. -

Genetic Manipulation for Inherited Neurodegenerative Diseases: Myth Or Reality? Patrick Yu-Wai-Man1,2,3
BJO Online First, published on August 1, 2016 as 10.1136/bjophthalmol-2015-308329 Review Br J Ophthalmol: first published as 10.1136/bjophthalmol-2015-308329 on 21 March 2016. Downloaded from Genetic manipulation for inherited neurodegenerative diseases: myth or reality? Patrick Yu-Wai-Man1,2,3 1Wellcome Trust Centre for ABSTRACT genetic manipulation seems an obvious solution as Mitochondrial Research, Rare genetic diseases affect about 7% of the general a radical means of correcting the primary patho- Institute of Genetic Medicine, Newcastle University, population and over 7000 distinct clinical syndromes logical process that contributes to neuronal dys- Newcastle upon Tyne, UK have been described with the majority being due to function and disease progression. 2Newcastle Eye Centre, Royal single gene defects. This review will provide a critical Victoria Infirmary, Newcastle overview of genetic strategies that are being pioneered upon Tyne, UK GENE THERAPY PARADIGMS 3 to halt or reverse disease progression in inherited ‘ NIHR Biomedical Research fi Gene therapy is a priori perfectly suited for single Centre at Moorfields Eye neurodegenerative diseases. This eld of research covers gene’ neurological disorders, but some additional Hospital and UCL Institute of a vast area and only the most promising treatment criteria need to be fulfilled, namely: (1) there must Ophthalmology, London, UK paradigms will be discussed with a particular focus on be a relatively clear understanding of the pathways inherited eye diseases, which have paved the way for Correspondence to contributing to neuronal loss to ensure selection of Dr Patrick Yu-Wai-Man, innovative gene therapy paradigms, and mitochondrial the most appropriate therapeutic strategy; (2) the Wellcome Trust Centre for diseases, which are currently generating a lot of debate natural history of the disease must be clearly Mitochondrial Research, centred on the bioethics of germline manipulation. -

Prevention Ol Childhood Bundness
Prevention ol childhood bUndness World Health- Orpalzatloa Geaen Prevention of childhood blindness Prevention of childhood blindness World Health Organization Geneva 1992 WHO Library Catalogumg in Publication Data Prevention of childhood blindness. r.Blmdness - in infancy & childhood 2.Biindness - prevention & control ISBN 92 4 156151 3 (NLM Classification: WW 276) The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. Applications and enquiries should be addressed to the Office of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available. © WORLD HEALTH ORGANIZATION 1992 Publications of the World Health Organization enjoy copyright protection in accordance with the provisions ofProtocol2 of the Umversal Copyright Convention. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of Its authorities, or concerning the delimitation of its frontiers or boundaries. The mention of specific companies or of certain manufacturers' products does not Imply that they are endorsed or recommended by the World Health Organization m preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. TYPESET IN INDIA PRINTED IN ENGLAND 91 /9088-Macmillan/Clays/GCW-s8oo Contents Preface vu Introduction I I.