Recent Advances in Oligonucleotide Therapeutics in Oncology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Phase II Study of the Novel Proteasome Inhibitor Bortezomib In
Memorial Sloan-Kettering Cance r Center IRB Protocol IRB#: 05-103 A(14) A Phase II Study of the Novel Proteas ome Inhibitor Bortezomib in Combination with Rituximab, Cyclophosphamide and Prednisone in Patients with Relapsed/Refractory I Indolent B-cell Lymphoproliferative Disorders and Mantle Cell Lymphoma (MCL) MSKCC THERAPEUTIC/DIAGNOSTIC PROTOCOL Principal Investigator: John Gerecitano, M.D., Ph.D. Co-Principal Carol Portlock, M.D. Investigator(s): IFormerly: A Phase I/II Study of the Nove l Proteasome Inhibitor Bortezomib in Combinati on with Rituximab, Cyclophosphamide and Prednisone in Patients with Relapsed/Refractory Indolent B-cell Lymphoproliferative Disorders and Mantle Cell Lymphoma (MCL) Amended: 07/25/12 Memorial Sloan-Kettering Cance r Center IRB Protocol IRB#: 05-103 A(14) Investigator(s): Paul Hamlin, M.D. Commack, NY Steven B. Horwitz, M.D. Philip Schulman, M.D. Alison Moskowitz, M.D. Stuart Lichtman, M.D Craig H. Moskowitz, M.D. Stefan Berger, M.D. Ariela Noy, M.D. Julie Fasano, M.D. M. Lia Palomba, M.D., Ph.D. John Fiore, M.D. Jonathan Schatz, M.D. Steven Sugarman, M.D David Straus, M.D. Frank Y. Tsai, M.D. Andrew D. Zelenetz, M.D., Ph.D. Matthew Matasar, M.D Rockville Center, NY Mark L. Heaney, M.D., Ph.D. Pamela Drullinksy, M.D Nicole Lamanna, M.D. Arlyn Apollo, M.D. Zoe Goldberg, M.D. Radiology Kenneth Ng, M.D. Otilia Dumitrescu, M.D. Tiffany Troso-Sandoval, M.D. Andrei Holodny, M.D. Sleepy Hollow, NY Nuclear Medicine Philip Caron, M.D. Heiko Schoder, M.D. Michelle Boyar, M.D. -

(12) United States Patent (10) Patent No.: US 8,580,814 B2 Adelman Et Al
USOO858O814B2 (12) United States Patent (10) Patent No.: US 8,580,814 B2 Adelman et al. (45) Date of Patent: *Nov. 12, 2013 (54) METHODS OF USING 5,528,823. A 6/1996 Rudy, Jr. et al. (+)-1,4-DIHYDRO-7-(3S4S)-3-METHOXY-4- 3.43wk A E. E. St.Ola tala (METHYLAMINO)-1-PYRROLIDINYL-4- 6,171,857 B1 1/2001 Hendrickson OXO-1-(2-THIAZOLYL)-1.8- 6,291643 B1 9/2001 Zou et al. NAPHTHYRIDNE-3-CARBOXYLIC ACID 6,570,002 B1 5/2003 Hardwicket al. FORTREATMENT OF CANCER 6,641,810 B2 11/2003 Gold 6,641,833 B2 * 1 1/2003 Dang ............................ 424/426 (75) Inventors: Daniel CAdelman, Redwood City, CA 6,696,4836,670,144 B2B1 12/20032/2004 CraigSingh et al. (US); Jeffrey A. Silverman, 6,723,734 B2 4/2004 Kim et al. Burlingame, CA (US) 7,211,562 B2 5/2007 Rosen et al. 7.989,468 B2 * 8/2011 Adelman et al. .............. 514,300 (73) Assignee: Sunesis Pharmaceuticals, Inc., South 3.9. A. S. idaran-Ghera Gh et al.1 San Francisco, CA (US) 2003/0232334 A1* 12/2003 Morris et al. ..................... 435/6 c - r 2004/0106605 A1 6/2004 Carboni et al. .... 514/226.8 (*) Notice: Subject to any disclaimer, the term of this 2004/0132825 A1* 7/2004 Bacopoulos et al. ......... 514/575 patent is extended or adjusted under 35 2005/0203120 A1 9, 2005 Adelman et al. U.S.C. 154(b) by 201 days. 2005/0215583 A1 9, 2005 Arkin et al. 2006, OO25437 A1 2/2006 Adelman et al. -
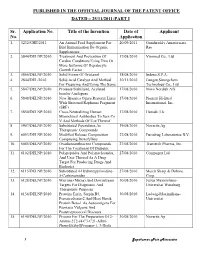
Published in the Official Journal of the Patent Office Dated – 25/11/2011-Part I
PUBLISHED IN THE OFFICIAL JOURNAL OF THE PATENT OFFICE DATED – 25/11/2011-PART I Sr. Application No. Title of the Invention Date of Applicant No. Application 1. 3232/CHE/2011 An Animal Feed Supplement For 20/09/2011 Gundareddy Amareswara Bird Immunization By Organic Rao Supplements 2. 5844/DELNP/2010 Treatment And Prevention Of 17/08/2010 Viromed Co., Ltd Cardiac Conditions Using Two Or More Isoforms Of Hepatocyte Growth Factor 3. 5866/DELNP/2010 Solid Forms Of Ortataxel 18/08/2010 Indena S.P.A. 4. 2844/DEL/2010 Solid Acid Catalyst And Method 30/11/2010 Jiangsu Sinorgchem For Preparing And Using The Same Technology Co., Ltd 5. 5847/DELNP/2010 Protease Stabilized, Acylated 17/08/2010 Novo Nordisk A/S Insulin Analogues 6. 5848/DELNP/2010 New Brassica Ogura Restorer Lines 17/08/2010 Pioneer Hi-Bred With Shotened Raphanus Fragment International, Inc. (SRF) 7. 5850/DELNP/2010 Cross-Neutralizing Human 17/08/2010 Humab, Llc Monoclonal Antibodies To Sars-Co V And Methods Of Use Thereof 8. 5907/DELNP/2010 Substituted Piperidines As 19/08/2010 Novartis Ag Therapeutic Compounds 9. 6093/DELNP/2010 Modified Release Composition 27/08/2010 Eurodrug Laboratories B.V. Comprising Doxofylline 10. 6085/DELNP/2010 Oxadiazoanthracene Compounds 27/08/2010 Transtech Pharma, Inc. For The Treatment Of Diabetes 11. 6102/DELNP/2010 Polypeptides And Polynucleotides, 27/08/2010 Compugen Ltd And Uses Thereof As A Drug Target For Producing Drugs And Biologics 12. 6115/DELNP/2010 Substituted 4-Hydroxypyrimidine- 27/08/2010 Merck Sharp & Dohme 5-Carboxamides Corp. 13. 6128/DELNP/2010 Microna (Mirna) And Downstream 30/08/2010 Julius Maximilians- Targets For Diagnostic And Universitat Wurzburg Therapeutic Purposes 14. -

Amyloid Seeding of Transthyretin by Ex Vivo Cardiac Fibrils and Its
Amyloid seeding of transthyretin by ex vivo cardiac PNAS PLUS fibrils and its inhibition Lorena Saelicesa,b,c,d, Kevin Chunga,b,c,d, Ji H. Leea,b,c,d, Whitaker Cohne, Julian P. Whiteleggee, Merrill D. Bensonf, and David S. Eisenberga,b,c,d,1 aHoward Hughes Medical Institute, University of California, Los Angeles, CA 90095; bUCLA-DOE, University of California, Los Angeles, CA 90095; cDepartment of Biological Chemistry, University of California, Los Angeles, CA 90095; dMolecular Biology Institute, University of California, Los Angeles, CA 90095; eNeuropsychiatric Institute (NPI)-Semel Institute, University of California, Los Angeles, CA 90024; and fDepartment of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, IN 46202 Contributed by David S. Eisenberg, April 18, 2018 (sent for review November 8, 2017; reviewed by Joel N. Buxbaum, Jeffery W. Kelly, and Gunilla T. Westermark) Each of the 30 human amyloid diseases is associated with the both full-length TTR fibrils and C-terminal TTR fragments. In aggregation of a particular precursor protein into amyloid fibrils. In type B cardiac ATTR, more distinct amyloid deposits made of transthyretin amyloidosis (ATTR), mutant or wild-type forms of the full-length TTR fibrils surround individual muscle cells. Although serum carrier protein transthyretin (TTR), synthesized and secreted the understanding of their clinical and pathological significance is by the liver, convert to amyloid fibrils deposited in the heart and incomplete, there is a clear distinction between subtypes: type A other organs. The current standard of care for hereditary ATTR is deposits display a higher capacity to recruit wild-type TTR (16). -

Opportunities and Challenges for Antisense Oligonucleotide Therapies
Received: 10 January 2020 Revised: 23 April 2020 Accepted: 8 May 2020 DOI: 10.1002/jimd.12251 REVIEW ARTICLE Opportunities and challenges for antisense oligonucleotide therapies Elsa C. Kuijper1 | Atze J. Bergsma2,3 | W.W.M. Pim Pijnappel2,3 | Annemieke Aartsma-Rus1 1Department of Human Genetics, Leiden University Medical Center, Leiden, The Abstract Netherlands Antisense oligonucleotide (AON) therapies involve short strands of modi- 2Department of Pediatrics, Center for fied nucleotides that target RNA in a sequence-specific manner, inducing Lysosomal and Metabolic Diseases, targeted protein knockdown or restoration. Currently, 10 AON therapies Erasmus Medical Center, Rotterdam, The Netherlands have been approved in the United States and Europe. Nucleotides are chem- 3Department of Clinical Genetics, Center ically modified to protect AONs from degradation, enhance bioavailability for Lysosomal and Metabolic Diseases, and increase RNA affinity. Whereas single stranded AONs can efficiently Erasmus Medical Center, Rotterdam, The Netherlands be delivered systemically, delivery of double stranded AONs requires capsulation in lipid nanoparticles or binding to a conjugate as the uptake Correspondence enhancing backbone is hidden in this conformation. With improved chem- Annemieke Aartsma-Rus, LUMC Postzone S4-P, Albinusdreef 2, 2333 ZA istry, delivery vehicles and conjugates, doses can be lowered, thereby reduc- Leiden, The Netherlands. ing the risk and occurrence of side effects. AONs can be used to knockdown Email: [email protected] or restore levels of protein. Knockdown can be achieved by single stranded Communicating Editor: Carla E. Hollak or double stranded AONs binding the RNA transcript and activating RNaseH-mediated and RISC-mediated degradation respectively. Transcript binding by AONs can also prevent translation, hence reducing protein levels. -

Preclinical Pharmacologic Evaluation of Pralatrexate and Romidepsin
Published OnlineFirst February 12, 2015; DOI: 10.1158/1078-0432.CCR-14-2249 Cancer Therapy: Preclinical Clinical Cancer Research Preclinical Pharmacologic Evaluation of Pralatrexate and Romidepsin Confirms Potent Synergy of the Combination in a Murine Model of Human T-cell Lymphoma Salvia Jain1, Xavier Jirau-Serrano2, Kelly M. Zullo2, Luigi Scotto2, Carmine F. Palermo3,4, Stephen A. Sastra3,4,5, Kenneth P. Olive3,4,5, Serge Cremers3, Tiffany Thomas3,YingWei6, Yuan Zhang6, Govind Bhagat3, Jennifer E. Amengual2, Changchun Deng2, Charles Karan8, Ronald Realubit8, Susan E. Bates9, and Owen A. O'Connor2 Abstract Purpose: T-cell lymphomas (TCL) are aggressive diseases, NOG mouse model of TCL were used to explore the in vitro and in which carry a poor prognosis. The emergence of new drugs for vivo activity of pralatrexate and romidepsin in combination. TCL has created a need to survey these agents in a rapid and Corresponding mass spectrometry–based pharmacokinetic and reproducible fashion, to prioritize combinations which should be immunohistochemistry-based pharmacodynamic analyses of prioritized for clinical study. Mouse models of TCL that can be xenograft tumors were performed to better understand a mech- used for screening novel agents and their combinations are anistic basis for the drug:drug interaction. lacking. Developments in noninvasive imaging modalities, such Results: In vitro, pralatrexate and romidepsin exhibited con- as surface bioluminescence (SBL) and three-dimensional ultra- centration-dependent synergism in combination against a panel sound (3D-US), are challenging conventional approaches in of TCL cell lines. In a NOG murine model of TCL, the combination xenograft modeling relying on caliper measurements. The recent of pralatrexate and romidepsin exhibited enhanced efficacy com- approval of pralatrexate and romidepsin creates an obvious pared with either drug alone across a spectrum of tumors using combination that could produce meaningful activity in TCL, complementary imaging modalities, such as SBL and 3D-US. -

EP Vantage Interview - Silence Hoping to Have Something to Shout About in 2009
December 22, 2008 EP Vantage Interview - Silence hoping to have something to shout about in 2009 Lisa Urquhart With its first product expected to go into the clinic potentially as early as January, Silence Therapeutics is hoping that 2009 will be year it has something to shout about and one that will reverse the alarming share price decline that has seen the company’s valuation slip from a high of over £170m in June 2007 to £21.6m today. Silence is one of a growing number of companies working in RNA interference (RNAi) and particularly short interfering RNA (siRNA), which work by selectively silencing or inactivating genes related to certain diseases. It is importantly one of only two companies that have composition of matter patent protection for their siRNA drugs. Speaking to EP Vantage, Iain Ross, chief executive of Silence, says: “It’s a big year coming up for us.” The group has spent most of the last 12 months strengthening its IP position, and next year comes the important move of the group’s lead candidate Atu027 into the clinic, an event Mr Ross says should take place in the first quarter of the year. Many expect it could be as soon as the end of January. Partnering focus What is less expected is the speed at which Mr Ross intends to partner the drug, which is being developed in solid tumours, with an emphasis on lung cancer. “We would be looking at doing something either at the end of 2009 or the beginning of 2010,” he says. Key to partnering discussions will be the drug demonstrating good safety data in a number of cancer patients, which could be the catalyst to starting talks with big pharma who have already started to ask about Atu027. -

Oregon Medicaid Pharmaceutical Services Prior Authorization Criteria
Oregon Medicaid Pharmaceutical Services Prior Authorization Criteria HEALTH SYSTEMS DIVISION Prior authorization (PA) criteria for fee-for-service prescriptions for Oregon Health Plan clients March 1, 2021 Contents Contents ................................................................................................................................................................ 2 Introduction........................................................................................................................................................... 7 About this guide ......................................................................................................................................... 7 How to use this guide ................................................................................................................................. 7 Administrative rules and supplemental information .................................................................................. 7 Update information............................................................................................................................................... 8 Effective March 1, 2021 ............................................................................................................................ 8 Substantive updates and new criteria ............................................................................................. 8 Clerical changes ............................................................................................................................ -

Medicines Not Reimbursed Through National Prices and Directly Commissioned by Nhs England V14 Changes to Version 13
MEDICINES NOT REIMBURSED THROUGH NATIONAL PRICES AND DIRECTLY COMMISSIONED BY NHS ENGLAND V14 CHANGES TO VERSION 13 PUBLISHED APRIL 2019 Changes to v13 Lines removed SUITABLE SPECIALIST FOR CENTRE SUITABLE FOR SHARED ONLY SHARED CARE CARE PRIOR (includng WITH PRIMARY BETWEEN STOPPING MONITORING/ AUDIT APPROVAL outreach CARE (IF DRUG NAME INDICATION COMMISSIONER PBR CATEGORY TA/POLICY STARTING CRITERIA SPECIALIST COMMENT CRITERIA REQUIREMENTS PROFORMA when SUPPORTED AND REQUIRED delivered as BY LOCAL SECONDARY part of a PRESCRIBING CARE VIA provider COMMITTEE) NETWORK network) MODEL PAEDIATRIC INDICATIONS (IN LINE WITH NHS ENGLAND AS PER ADULT TA'S (TA195, TA373, ABATACEPT NHS ENGLAND CYTOKINE MODULATORS NICE NICE NICE √ MEDICINES FOR CHILDREN TA375) POLICY) Now covered by comment 8 PAEDIATRIC INDICATIONS (IN AS PER TA455 OR ADULT TA'S (TA130 LINE WITH NHS ENGLAND (replaced by TA375), TA143 (repalced by ADALIMUMAB NHS ENGLAND CYTOKINE MODULATORS NICE NICE NICE AUDIT √ √ MEDICINES FOR CHILDREN TA383), TA146, TA187, TA199, TA329, POLICY) TA392, TA460) Now covered by comment 8 ALBUTREPENONACOG ALFA HAEMOPHILIA B PRODUCTS ON CMU Blood factor products simplified NHS ENGLAND BLOOD-RELATED PRODUCTS SSC 1652 SSC 1652 SSC 1652 √ FRAMEWORK to blood factor product VIII etc PAEDIATRIC INDICATIONS (IN LINE WITH NHS ENGLAND AS PER IFR AS PER IFR ANAKINRA NHS ENGLAND CYTOKINE MODULATORS NOT ROUTINELY COMMISSIONED AS PER IFR APPROVAL √ MEDICINES FOR CHILDREN APPROVAL APPROVAL POLICY) Now covered by comment 8 AS PER BCSH GUIDELINES ANTIHAEMOPHILIC FACTOR/VON -

RNA Pull-Down Procedure to Identify RNA Targets of a Long Non-Coding
RNA Pull-down Procedure to Identify RNA Targets of a Long Non-coding RNA Manon Torres, Denis Becquet, Séverine Guillen, Bénédicte Boyer, Mathias Moreno, Marie-Pierre Blanchard, Jean-Louis Franc, Anne-Marie François-Bellan To cite this version: Manon Torres, Denis Becquet, Séverine Guillen, Bénédicte Boyer, Mathias Moreno, et al.. RNA Pull-down Procedure to Identify RNA Targets of a Long Non-coding RNA. Journal of visualized experiments : JoVE, JoVE, 2018, 10.3791/57379. hal-02093065 HAL Id: hal-02093065 https://hal-amu.archives-ouvertes.fr/hal-02093065 Submitted on 20 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License Journal of Visualized Experiments www.jove.com Video Article RNA Pull-down Procedure to Identify RNA Targets of a Long Non-coding RNA Manon Torres1, Denis Becquet1, Séverine Guillen1, Bénédicte Boyer1, Mathias Moreno1, Marie-Pierre Blanchard2, Jean-Louis Franc1, Anne- Marie François-Bellan1 1 CNRS, CRN2M-UMR7286, Faculté -

Advances in Oligonucleotide Drug Delivery
REVIEWS Advances in oligonucleotide drug delivery Thomas C. Roberts 1,2 ✉ , Robert Langer 3 and Matthew J. A. Wood 1,2 ✉ Abstract | Oligonucleotides can be used to modulate gene expression via a range of processes including RNAi, target degradation by RNase H-mediated cleavage, splicing modulation, non-coding RNA inhibition, gene activation and programmed gene editing. As such, these molecules have potential therapeutic applications for myriad indications, with several oligonucleotide drugs recently gaining approval. However, despite recent technological advances, achieving efficient oligonucleotide delivery, particularly to extrahepatic tissues, remains a major translational limitation. Here, we provide an overview of oligonucleotide-based drug platforms, focusing on key approaches — including chemical modification, bioconjugation and the use of nanocarriers — which aim to address the delivery challenge. Oligonucleotides are nucleic acid polymers with the In addition to their ability to recognize specific tar- potential to treat or manage a wide range of diseases. get sequences via complementary base pairing, nucleic Although the majority of oligonucleotide therapeutics acids can also interact with proteins through the for- have focused on gene silencing, other strategies are being mation of three-dimensional secondary structures — a pursued, including splice modulation and gene activa- property that is also being exploited therapeutically. For tion, expanding the range of possible targets beyond example, nucleic acid aptamers are structured -

Guide for Morpholino Users: Toward Therapeutics
Open Access Journal of Drug Discovery, Development and Delivery Special Article - Antisense Drug Research and Development Guide for Morpholino Users: Toward Therapeutics Moulton JD* Gene Tools, LLC, USA Abstract *Corresponding author: Moulton JD, Gene Tools, Morpholino oligos are uncharged molecules for blocking sites on RNA. They LLC, 1001 Summerton Way, Philomath, Oregon 97370, are specific, soluble, non-toxic, stable, and effective antisense reagents suitable USA for development as therapeutics and currently in clinical trials. They are very versatile, targeting a wide range of RNA targets for outcomes such as blocking Received: January 28, 2016; Accepted: April 29, 2016; translation, modifying splicing of pre-mRNA, inhibiting miRNA maturation and Published: May 03, 2016 activity, as well as less common biological targets and diagnostic applications. Solutions have been developed for delivery into a range of cultured cells, embryos and adult animals; with development of a non-toxic and effective system for systemic delivery, Morpholinos have potential for broad therapeutic development targeting pathogens and genetic disorders. Keywords: Splicing; Duchenne muscular dystrophy; Phosphorodiamidate morpholino oligos; Internal ribosome entry site; Nonsense-mediated decay Morpholinos: Research Applications, the transcript from miRNA regulation; Therapeutic Promise • Block regulatory proteins from binding to RNA, shifting Morpholino oligos bind to complementary sequences of RNA alternative splicing; and get in the way of processes. Morpholino oligos are commonly • Block association of RNAs with cytoskeletal motor protein used to prevent a particular protein from being made in an organism complexes, preventing RNA translocation; or cell culture. Morpholinos are not the only tool used for this: a protein’s synthesis can be inhibited by altering DNA to make a null • Inhibit poly-A tailing of pre-mRNA; mutant (called a gene knockout) or by interrupting processes on RNA • Trigger frame shifts at slippery sequences; (called a gene knockdown).