Return of a Lost Structure in the Evolution of Felid Dentition Revisited: a Devoevo Perspective on the Irreversibility of Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
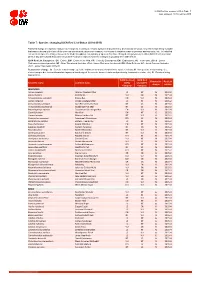
Table 7: Species Changing IUCN Red List Status (2014-2015)
IUCN Red List version 2015.4: Table 7 Last Updated: 19 November 2015 Table 7: Species changing IUCN Red List Status (2014-2015) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2014 (IUCN Red List version 2014.3) and 2015 (IUCN Red List version 2015-4) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered, EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2014) List (2015) change version Category Category MAMMALS Aonyx capensis African Clawless Otter LC NT N 2015-2 Ailurus fulgens Red Panda VU EN N 2015-4 -

Current Issue
Current Issue Cat Project of the Month - September 2005 The IUCN/SSC Cat Specialist Group's website (www.catsg.org) presents each month a different cat conservation project. Members of the Cat Specialist Group are encouraged to submit a short description of interesting projects For application use this standardised form (an editable word document) Pampas cat ecology and conservation in the Brazilian grasslands The main goal of the study on pampas cats in the Brazilian grasslands is to rise information on the ecology of the species, including data on diseases, genetics and threats. Leandro Silveira, Anah T. A. Jácomo and Mariana Malzoni Furtado Leandro Silveira (photo) is a wildlife biologist (PhD). He is the project leader of the Carnovire Community Project at Emas Naitonal Park and is the president of the Jaguar Conservation Fund. Leandro has been a member of the Cat SG since 2002. Since 2003, he is conduction a study on the papas cat (Photo Jaugar Conservation Fund) [email protected] submitted: 23/09/2005 The pampas cat (Oncifelis colocolo) is widely distributed in a variety of South American habitats. It ranges from the forested Andean slopes of Ecuador, Peru and Bolívia, cloud forest in central Chile, Paraguayan chaco, open woodlands of central, western, northeastern and southern Brazil, the Argentine pampas, southern Patagonia, and the pampas of Uruguay (Cabrera 1957, Vieira, 1955; Ximenez, 1961). In Brazil it is restricted to open habitats and it is found in the Cerrado of central Brazil, the Pantanal of western Mato Grosso do Sul, and in southern open grassland and mangrove habitats. -

Pygmy Hog – 1 Southern Ningaul – 16 Kowari – 9 Finlayson's Squirrel
Pygmy Hog – 1 Habitat: Diurnal/Nocturnal Defense: Size Southern Ningaul – 16 Habitat: Diurnal/Nocturnal Defense: Size Kowari – 9 Habitat: Diurnal/Nocturnal Defense: Size Finlayson’s Squirrel – 8 Habitat: Diurnal/Nocturnal Defense: Size Kodkod – 5 Habitat: Diurnal/Nocturnal Defense: Size Least Chipmunk – 12 Habitat: Diurnal/Nocturnal Defense: Size Tree Hyrax – 4 Habitat: Diurnal/Nocturnal Defense: Size Bank Vole – 13 Habitat: Diurnal/Nocturnal Defense: Size Island Fox – 6 Habitat: Diurnal/Nocturnal Defense: Size Gray-bellied Caenolestid – 11 Habitat: Diurnal/Nocturnal Defense: Size Raccoon Dog – 3 Habitat: Diurnal/Nocturnal Defense: Size Northern Short-Tailed Shrew – 14 Habitat: Diurnal/Nocturnal Defense: Size Southern African Hedgehog - 7 Habitat: Diurnal/Nocturnal Defense: Size Collard Pika - 10 Habitat: Diurnal/Nocturnal Defense: Size Pudu – 2 Habitat: Diurnal/Nocturnal Defense: Size Seba’s Short-tailed Bat – 15 Habitat: Diurnal/Nocturnal Defense: Size Pygmy Spotted Skunk - 16 Habitat: Diurnal/Nocturnal Defense: Size Grandidier’s “Mongoose” - 16 Habitat: Diurnal/Nocturnal Defense: Size Sloth Bear - 1 Habitat: Diurnal/Nocturnal Defense: Size Spotted Linsing - 9 Habitat: Diurnal/Nocturnal Defense: Size Red Panda - 8 Habitat: Diurnal/Nocturnal Defense: Size European Badger – 5 Habitat: Diurnal/Nocturnal Defense: Size Giant Forest Genet – 12 Habitat: Diurnal/Nocturnal Defense: Size African Civet – 4 Habitat: Diurnal/Nocturnal Defense: Size Kinkajou – 13 Habitat: Diurnal/Nocturnal Defense: Size Fossa – 6 Habitat: Diurnal/Nocturnal Defense: -

Dental and Temporomandibular Joint Pathology of the Kit Fox (Vulpes Macrotis)
Author's Personal Copy J. Comp. Path. 2019, Vol. 167, 60e72 Available online at www.sciencedirect.com ScienceDirect www.elsevier.com/locate/jcpa DISEASE IN WILDLIFE OR EXOTIC SPECIES Dental and Temporomandibular Joint Pathology of the Kit Fox (Vulpes macrotis) N. Yanagisawa*, R. E. Wilson*, P. H. Kass† and F. J. M. Verstraete* *Department of Surgical and Radiological Sciences and † Department of Population Health and Reproduction, School of Veterinary Medicine, University of California, Davis, California, USA Summary Skull specimens from 836 kit foxes (Vulpes macrotis) were examined macroscopically according to predefined criteria; 559 specimens were included in this study. The study group consisted of 248 (44.4%) females, 267 (47.8%) males and 44 (7.9%) specimens of unknown sex; 128 (22.9%) skulls were from young adults and 431 (77.1%) were from adults. Of the 23,478 possible teeth, 21,883 teeth (93.2%) were present for examina- tion, 45 (1.9%) were absent congenitally, 405 (1.7%) were acquired losses and 1,145 (4.9%) were missing ar- tefactually. No persistent deciduous teeth were observed. Eight (0.04%) supernumerary teeth were found in seven (1.3%) specimens and 13 (0.06%) teeth from 12 (2.1%) specimens were malformed. Root number vari- ation was present in 20.3% (403/1,984) of the present maxillary and mandibular first premolar teeth. Eleven (2.0%) foxes had lesions consistent with enamel hypoplasia and 77 (13.8%) had fenestrations in the maxillary alveolar bone. Periodontitis and attrition/abrasion affected the majority of foxes (71.6% and 90.5%, respec- tively). -

Oncilla 1 Oncilla
Oncilla 1 Oncilla Tiger Cat redirects here. For the Tom and Jerry Tales episode, see Tiger Cat (Tom and Jerry Tales). Oncilla[1] Conservation status [2] Vulnerable (IUCN 3.1) Scientific classification Kingdom: Animalia Phylum: Chordata Class: Mammalia Order: Carnivora Family: Felidae Genus: Leopardus Species: L. tigrinus Binomial name Leopardus tigrinus (Schreber, 1775) Oncilla 2 Oncilla range Synonyms Oncifelis tigrinus Felis tigrina The Oncilla (Leopardus tigrinus), also known as the Little Spotted Cat, Tigrillo, Cunaguaro or Tiger Cat, is a small spotted felid found in the tropical rainforests of Central and South America. It is a close relative of the Ocelot and the Margay, and has a rich ochre coat, spotted with black rosettes. The Oncilla is a nocturnal animal that hunts rodents and birds.[3] Appearance The Oncilla resembles the Margay and the Ocelot,[4] but is smaller, with a slender build and narrower muzzle. It grows to 38 to 59 centimetres (15 to 23 in) long, plus a 20 to 42 centimetres (7.9 to 17 in) tail.[5] While this is somewhat longer than the average domestic cat, Leopardus tigrinus is generally lighter, weighing 1.5 to 3 kilograms (3.3 to 6.6 lb).[6] The fur is thick and soft, ranging from light brown to dark ochre, with numerous dark rosettes across the back and flanks. The underside is pale with dark spots and the tail is ringed. The backs of the ears are black with bold ocelli.[4] The rosettes are black or brown, open in the center, and irregularly shaped.[7] The legs have medium-sized spots tapering to smaller spots near the paws.[7] This coloration helps the oncilla blend in with the mottled sunlight of the tropical forest understory. -

Eurasian Lynx – Your Essential Brief
Eurasian lynx – Your essential brief Background Q: Are lynx native to Britain? A: Based on archaeological evidence, the range of the Eurasian lynx (Lynx lynx) included Britain until at least 1,300 years ago. It is difficult to be precise about when or why lynx became extinct here, but it was almost certainly related to human activity – deforestation removed their preferred habitat, and also that of their prey, thus reducing prey availability. These declines in prey species may have been exacerbated by human hunting. Q: Where do they live now? A: Across Europe, Scandinavia, Russia, northern China and Southeast Asia. The range used to include other areas of Western Europe, including Britain, where they are no longer present. Q: How many are there? A: There are thought to be around 50,000 in the world, of which 9,000 – 10,000 live in Europe. They are considered to be a species of least concern by the IUCN. Modern range of the Eurasian lynx Q: How big are they? A: Lynx are on average around 1m in length, 75cm tall and around 20kg, with the males being slightly larger than the females. They can live to 15 years old, but this is rare in the wild. Q: What do they eat? A: The preferred prey of the lynx are the smaller deer species, primarily the roe deer. Lynx may also prey upon other deer species, including chamois, sika deer, smaller red deer, muntjac and fallow deer. Q: Do they eat other things? A: Yes. Lynx prey on many other species when their preferred prey is scarce, including rabbits, hares, foxes, wildcats, squirrel, pine marten, domestic pets, sheep, goats and reared gamebirds. -

Status of Large Carnivores in Serbia
Status of large carnivores in Serbia Duško Ćirović Faculty of Biology University of Belgrade, Belgrade Status and threats of large carnivores in Serbia LC have differend distribution, status and population trends Gray wolf Eurasian Linx Brown Bear (Canis lupus) (Lynx lynx ) (Ursus arctos) Distribution of Brown Bear in Serbia Carpathian Dinaric-Pindos East Balkan Population status of Brown Bear in Serbia Dinaric-Pindos: Distribution 10000 km2 N=100-120 Population increase Range expansion Carpathian East Balkan: Distribution 1400 km2 Dinaric-Pindos N= a few East Balkan Population trend: unknown Carpathian: Distribution 8200 km2 N=8±2 Population stable Legal status of Brown Bear in Serbia According Law on Protection of Nature and the Law on Game and Hunting brown bear in Serbia is strictly protected species. He is under the centralized jurisdiction of the Ministry of Environmental Protection Treats of Brown Bear in Serbia Intensive forestry practice and infrastructure development . Illegal killing Low acceptance due to fear for personal safety Distribution of Gray wolf in Serbia Carpathian Dinaric-Pindos East Balkan Population status of Gray wolf in Serbia Dinaric-Balkan: 2 Carpathian Distribution cca 43500 km N=800-900 Population - stabile/slight increasingly Dinaric Range - slight expansion Carpathian: Distribution 480 km2 (was) Population – a few Population status of Gray wolf in Serbia Carpathian population is still undefined Carpathian Peri-Carpathian Legal status of Gray wolf in Serbia According the Law on Game and Hunting the gray wolf in majority pars of its distribution (south from Sava and Danube rivers) is game species with closing season from April 15th to July 1st. -

Controlled Animals
Environment and Sustainable Resource Development Fish and Wildlife Policy Division Controlled Animals Wildlife Regulation, Schedule 5, Part 1-4: Controlled Animals Subject to the Wildlife Act, a person must not be in possession of a wildlife or controlled animal unless authorized by a permit to do so, the animal was lawfully acquired, was lawfully exported from a jurisdiction outside of Alberta and was lawfully imported into Alberta. NOTES: 1 Animals listed in this Schedule, as a general rule, are described in the left hand column by reference to common or descriptive names and in the right hand column by reference to scientific names. But, in the event of any conflict as to the kind of animals that are listed, a scientific name in the right hand column prevails over the corresponding common or descriptive name in the left hand column. 2 Also included in this Schedule is any animal that is the hybrid offspring resulting from the crossing, whether before or after the commencement of this Schedule, of 2 animals at least one of which is or was an animal of a kind that is a controlled animal by virtue of this Schedule. 3 This Schedule excludes all wildlife animals, and therefore if a wildlife animal would, but for this Note, be included in this Schedule, it is hereby excluded from being a controlled animal. Part 1 Mammals (Class Mammalia) 1. AMERICAN OPOSSUMS (Family Didelphidae) Virginia Opossum Didelphis virginiana 2. SHREWS (Family Soricidae) Long-tailed Shrews Genus Sorex Arboreal Brown-toothed Shrew Episoriculus macrurus North American Least Shrew Cryptotis parva Old World Water Shrews Genus Neomys Ussuri White-toothed Shrew Crocidura lasiura Greater White-toothed Shrew Crocidura russula Siberian Shrew Crocidura sibirica Piebald Shrew Diplomesodon pulchellum 3. -

First Record of Hose's Civet Diplogale Hosei from Indonesia
First record of Hose’s Civet Diplogale hosei from Indonesia, and records of other carnivores in the Schwaner Mountains, Central Kalimantan, Indonesia Hiromitsu SAMEJIMA1 and Gono SEMIADI2 Abstract One of the least-recorded carnivores in Borneo, Hose’s Civet Diplogale hosei , was filmed twice in a logging concession, the Katingan–Seruyan Block of Sari Bumi Kusuma Corporation, in the Schwaner Mountains, upper Seruyan River catchment, Central Kalimantan. This, the first record of this species in Indonesia, is about 500 km southwest of its previously known distribution (northern Borneo: Sarawak, Sabah and Brunei). Filmed at 325The m a.s.l., IUCN these Red List records of Threatened are below Species the previously known altitudinal range (450–1,800Prionailurus m). This preliminary planiceps survey forPardofelis medium badia and large and Otter mammals, Civet Cynogalerunning 100bennettii camera-traps in 10 plots for one (Bandedyear, identified Civet Hemigalus in this concession derbyanus 17 carnivores, Arctictis including, binturong on Neofelis diardi, three Endangered Pardofe species- lis(Flat-headed marmorata Cat and Sun Bear Helarctos malayanus, Bay Cat . ) and six Vulnerable species , Binturong , Sunda Clouded Leopard , Marbled Cat Keywords Cynogale bennettii, as well, Pardofelis as Hose’s badia Civet), Prionailurus planiceps Catatan: PertamaBorneo, camera-trapping, mengenai Musang Gunung Diplogale hosei di Indonesia, serta, sustainable karnivora forest management lainnya di daerah Pegunungan Schwaner, Kalimantan Tengah Abstrak Diplogale hosei Salah satu jenis karnivora yang jarang dijumpai di Borneo, Musang Gunung, , telah terekam dua kali di daerah- konsesi hutan Blok Katingan–Seruyan- PT. Sari Bumi Kusuma, Pegunungan Schwaner, di sekitar hulu Sungai Seruya, Kalimantan Tengah. Ini merupakan catatan pertama spesies tersebut terdapat di Indonesia, sekitar 500 km dari batas sebaran yang diketa hui saat ini (Sarawak, Sabah, Brunei). -

The Leopardus Tigrinus Is One of the Smallest Wild Cats in South America; and the Smallest Cat in Brazil (Oliveira-Santos Et Al
Mckenzie Brocker Conservation Biology David Stokes 20 February 2014 Leopardus Tigrinus Description: The Leopardus tigrinus is one of the smallest wild cats in South America; and the smallest cat in Brazil (Oliveira-Santos et al. 2012). L. tigrinus is roughly the size of a domestic house cat, with its weight ranging from 1.8-3.4 kg (Silva-Pereira 2010). The average body length is 710 millimeters and the cat’s tail is roughly one-third of its body length averaging 250 millimeters in length. Males tend to be slightly larger than the females (Gardner 1971). The species’ coat is of a yellowish-brown or ochre coloration patterned prominently with open rosettes (Trigo et al. 2013). Cases of melanism, or dark pigmentation, have been reported but are not as common (Oliveira-Santos et al 2012). These characteristics spots are what give the L. tigrinus its common names of little spotted cat, little tiger cat, tigrina, tigrillo, and oncilla. The names tigrillo, little tiger cat, and little spotted cat are sometimes used interchangeably with other small Neotropical cats species which can lead to confusion. The species is closely related to other feline species with overlapping habitat areas and similar colorations; namely, the ocelot, Leopardus pardalis, the margay, Leopardus weidii, Geoffroys cat, Leopardus geoffroyi, and the pampas cat, Leopardus colocolo (Trigo et al. 2013). Distribution: The L. tigrinus is reported to have a wide distribution from as far north as Costa Rica to as far south as Northern Argentina. However, its exact distribution is still under study, as there have been few reports of occurrences in Central America. -

An Educator's Resource to Texas Mammal Skulls and Skins
E4H-014 11/17 An Educator’s Resource to Texas Mammal Skulls and Skins for use in 4-H Wildlife Programs and FFA Wildlife Career Development Events By, Denise Harmel-Garza Program Coordinator I, Texas A&M AgriLife Extension Service, 4-H Photographer and coauthor, Audrey Sepulveda M.Ed. Agricultural Leadership, Education and Communications, Texas A&M University College Station, Texas 2017 “A special thanks to the Biodiversity Research and Teaching Collections at Texas A&M University for providing access to their specimens.” Texas A&M AgriLife Extension provides equal opportunities in its programs and employment to all persons, regardless of race, color, sex, religion, national origin, disability, age, genetic information, veteran status, sexual orientation, or gender identity. The Texas A&M University System, U.S. Department of Agriculture, and the County Commissioners Courts of Texas Cooperating. Introduction Texas youth that participate in wildlife programs may be asked to identify a skull, skin, scat, tracks, etc. of an animal. Usually, educators must find this information and assemble pictures of skulls and skins from various sources. They also must ensure that what they find is relevant and accurate. Buying skulls and skins to represent all Texas mammals is costly. Most educators cannot afford them, and if they can, maintaining these collections over time is problematic. This study resource will reduce the time teachers across the state need to spend searching for information and allow them more time for presenting the material to their students. This identification guide gives teachers and students easy access to information that is accurate and valuable for learning to identify Texas mammals. -

Threats from Human Land Use and Climate Change to the Kodkod Leopardus Guigna
Assessing the distribution of a Vulnerable felid species: threats from human land use and climate change to the kodkod Leopardus guigna G RIET A.E. CUYCKENS,MIRIAM M. MORALES and M ARCELO F. TOGNELLI Abstract Climate change and habitat fragmentation are Introduction considered key pressures on biodiversity, and mammalian carnivores with a limited geographical distribution are limate change and destruction of natural habitats particularly vulnerable. The kodkod Leopardus guigna,a Cthrough human activities are major threats to terres- small felid endemic to the temperate forests of southern trial biodiversity. These pressures have led to substantial ’ Chile and Argentina, has the smallest geographical range range contractions and species extinctions (Parmesan & 2003 2003 2004 of any New World felid. Although the species occurs in Yohe, ; Root et al., ; Opdam & Wascher, ), 2008 protected areas in both countries, it is not known how well including for mammals (Schipper et al., ), and species 1991 these areas protect the kodkod either currently or under at high altitudes are particularly vulnerable (Innes, ; 1997 2003 climate change scenarios. We used species distribution Diaz & Bradley, ; Burns et al., ; Thomas et al., 2004 models and spatial analyses to assess the distribution of the ). Knowledge of the geographical distribution of species 2000 kodkod, examining the effects of changes in human land use is essential to assess these threats (Regan et al., ; Conrad 2006 and future climate change. We also assessed the species’ et al., ) but data on the distribution of rare species is 2006 2009 present representation in protected areas and in light of limited (Hernandez et al., ; Thorn et al., ).