Mouse Trip4 Conditional Knockout Project (CRISPR/Cas9)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Follow-Up of Loci from the International Genomics of Alzheimer’S Disease Project Identifies TRIP4 As a Novel Susceptibility Gene
OPEN Citation: Transl Psychiatry (2014) 4, e358; doi:10.1038/tp.2014.2 © 2014 Macmillan Publishers Limited All rights reserved 2158-3188/14 www.nature.com/tp ORIGINAL ARTICLE Follow-up of loci from the International Genomics of Alzheimer’s Disease Project identifies TRIP4 as a novel susceptibility gene A Ruiz1,32, S Heilmann2,3,32, T Becker4,5,32, I Hernández1, H Wagner6, M Thelen6, A Mauleón1, M Rosende-Roca1, C Bellenguez7,8,9, JC Bis10, D Harold11, A Gerrish11, R Sims11, O Sotolongo-Grau1, A Espinosa1, M Alegret1, JL Arrieta12, A Lacour4, M Leber4, J Becker6, A Lafuente1, S Ruiz1, L Vargas1, O Rodríguez1, G Ortega1, M-A Dominguez1, IGAP33, R Mayeux13,14, JL Haines15,16, MA Pericak-Vance17,18, LA Farrer19,20,21,22,23, GD Schellenberg24, V Chouraki23, LJ Launer25, C van Duijn26,27,28, S Seshadri23, C Antúnez29, MM Breteler4, M Serrano-Ríos30, F Jessen4,6, L Tárraga1, MM Nöthen2,3, W Maier4,6, M Boada1,31 and A Ramírez2,6 To follow-up loci discovered by the International Genomics of Alzheimer’s Disease Project, we attempted independent replication of 19 single nucleotide polymorphisms (SNPs) in a large Spanish sample (Fundació ACE data set; 1808 patients and 2564 controls). Our results corroborate association with four SNPs located in the genes INPP5D, MEF2C, ZCWPW1 and FERMT2, respectively. Of these, ZCWPW1 was the only SNP to withstand correction for multiple testing (P = 0.000655). Furthermore, we identify TRIP4 (rs74615166) as a novel genome-wide significant locus for Alzheimer’s disease risk (odds ratio = 1.31; confidence interval 95% (1.19–1.44); P = 9.74 × 10−9). -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Systems Biology Evaluation of Immune Responses Induced by Human Host Defence Peptide LL-37 in Mononuclear Cellsw
PAPER www.rsc.org/molecularbiosystems | Molecular BioSystems Systems biology evaluation of immune responses induced by human host defence peptide LL-37 in mononuclear cellsw Neeloffer Mookherjee,za Pamela Hamill,a Jennifer Gardy,a Darren Blimkie,b Reza Falsafi,a Avinash Chikatamarla,a David J. Arenillas,c Silvana Doria,a Tobias R. Kollmannb and Robert E. W. Hancock*a Received 7th August 2008, Accepted 29th January 2009 First published as an Advance Article on the web 19th March 2009 DOI: 10.1039/b813787k The immune system is very complex, it involves the integrated regulation and expression of hundreds of proteins. To understand in greater detail how the human host defence immunomodulatory peptide LL-37 interacts with innate immunity, a systems approach was pursued. Polychromatic flow cytometry was employed to demonstrate that within human peripheral blood mononuclear cells, CD14+ monocytes, myeloid and plasmocytoid dendritic cells and T- and B-lymphocytes, all responded to LL-37, with the differential production of intracellular cytokines. Microarray analyses with CD14+ monocytes indicated the differential expression of 475 genes in response to stimulation with LL-37. To understand this complex response, bioinformatic interrogation, using InnateDB, of the gene ontology, signalling pathways and transcription factor binding sites was undertaken. Activation of the IkBa/NFkB, mitogen- activated protein kinases p38, ERK1/2 and JNK, and PI3K signalling pathways in response to LL-37 was demonstrated by pathway and ontology over-representation analyses, and confirmed experimentally by inhibitor studies. Computational analysis of the predicted transcription factor binding sites upstream of the genes that were regulated by LL-37 predicted the involvement of several transcription factors including NFkB and five novel factors, AP-1, AP-2, SP-1, E2F1, and EGR, which were experimentally confirmed to respond to LL-37 by performing transcription factor array studies on nuclear extracts from LL-37 treated mononuclear cells. -

A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita
G C A T T A C G G C A T genes Article A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita Xenia Latypova 1, Stefan Giovanni Creadore 2, Noémi Dahan-Oliel 3,4, Anxhela Gjyshi Gustafson 2, Steven Wei-Hung Hwang 5, Tanya Bedard 6, Kamran Shazand 2, Harold J. P. van Bosse 5 , Philip F. Giampietro 7,* and Klaus Dieterich 8,* 1 Grenoble Institut Neurosciences, Université Grenoble Alpes, Inserm, U1216, CHU Grenoble Alpes, 38000 Grenoble, France; [email protected] 2 Shriners Hospitals for Children Headquarters, Tampa, FL 33607, USA; [email protected] (S.G.C.); [email protected] (A.G.G.); [email protected] (K.S.) 3 Shriners Hospitals for Children, Montreal, QC H4A 0A9, Canada; [email protected] 4 School of Physical & Occupational Therapy, Faculty of Medicine and Health Sciences, McGill University, Montreal, QC H3G 2M1, Canada 5 Shriners Hospitals for Children, Philadelphia, PA 19140, USA; [email protected] (S.W.-H.H.); [email protected] (H.J.P.v.B.) 6 Alberta Congenital Anomalies Surveillance System, Alberta Health Services, Edmonton, AB T5J 3E4, Canada; [email protected] 7 Department of Pediatrics, University of Illinois-Chicago, Chicago, IL 60607, USA 8 Institut of Advanced Biosciences, Université Grenoble Alpes, Inserm, U1209, CHU Grenoble Alpes, 38000 Grenoble, France * Correspondence: [email protected] (P.F.G.); [email protected] (K.D.) Citation: Latypova, X.; Creadore, S.G.; Dahan-Oliel, N.; Gustafson, Abstract: Arthrogryposis multiplex congenita (AMC) describes a group of conditions characterized A.G.; Wei-Hung Hwang, S.; Bedard, by the presence of non-progressive congenital contractures in multiple body areas. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Non‑Small‑Cell Lung Cancer Pathological Subtype‑Related Gene
356 MOLECULAR AND CLINICAL ONCOLOGY 8: 356-361, 2018 Non‑small‑cell lung cancer pathological subtype‑related gene selection and bioinformatics analysis based on gene expression profiles JIANGPENG CHEN1, XIAOQI DONG2, XUN LEI1, YINYIN XIA1, QING ZENG1, PING QUE1, XIAOYAN WEN1, SHAN HU1 and BIN PENG1 1School of Public Health and Management, Chongqing Medical University, Chongqing 400016; 2Department of Respiratory Diseases, The First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang 310003, P.R. China Received March 21, 2016; Accepted November 21, 2017 DOI: 10.3892/mco.2017.1516 Abstract. Lung cancer is one of the most common malignant Introduction diseases and a major threat to public health on a global scale. Non-small-cell lung cancer (NSCLC) has a higher degree of Lung cancer is one of the most common malignant diseases malignancy and a lower 5-year survival rate compared with that and a major threat to public health on a global scale. The main of small-cell lung cancer. NSCLC may be mainly divided into types of lung cancer are small-cell lung cancer (SCLC) and non- two pathological subtypes, adenocarcinoma and squamous cell small-cell lung cancer (NSCLC). NSCLC has a higher degree carcinoma. The aim of the present study was to identify disease of malignancy and a lower 5-year survival rate compared with genes based on the gene expression profile and the shortest path SCLC, and may be divided into two major histopathological analysis of weighted functional protein association networks subtypes, namely adenocarcinoma (ADC) and squamous cell with the existing protein-protein interaction data from the Search carcinoma (SCC). -

A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: a Simple System for Complex, Heterogeneous Diseases
International Journal of Molecular Sciences Review A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: A Simple System for Complex, Heterogeneous Diseases Weronika Rzepnikowska 1, Joanna Kaminska 2 , Dagmara Kabzi ´nska 1 , Katarzyna Bini˛eda 1 and Andrzej Kocha ´nski 1,* 1 Neuromuscular Unit, Mossakowski Medical Research Centre Polish Academy of Sciences, 02-106 Warsaw, Poland; [email protected] (W.R.); [email protected] (D.K.); [email protected] (K.B.) 2 Institute of Biochemistry and Biophysics Polish Academy of Sciences, 02-106 Warsaw, Poland; [email protected] * Correspondence: [email protected] Received: 19 May 2020; Accepted: 15 June 2020; Published: 16 June 2020 Abstract: Charcot–Marie–Tooth (CMT) disease encompasses a group of rare disorders that are characterized by similar clinical manifestations and a high genetic heterogeneity. Such excessive diversity presents many problems. Firstly, it makes a proper genetic diagnosis much more difficult and, even when using the most advanced tools, does not guarantee that the cause of the disease will be revealed. Secondly, the molecular mechanisms underlying the observed symptoms are extremely diverse and are probably different for most of the disease subtypes. Finally, there is no possibility of finding one efficient cure for all, or even the majority of CMT diseases. Every subtype of CMT needs an individual approach backed up by its own research field. Thus, it is little surprise that our knowledge of CMT disease as a whole is selective and therapeutic approaches are limited. There is an urgent need to develop new CMT models to fill the gaps. -
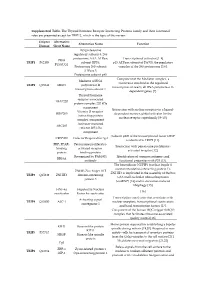
Supplemental Table. the Thyroid Hormone Receptor Interacting
Supplemental Table. The Thyroid hormone Receptor Interacting Proteins family and their functional roles are presented except for TRIP12, which is the topic of this review. Uniprot Alternative Alternative Name Function Human Short Name 26S proteasome regulatory subunit 8, 26S proteasome AAA-ATPase Transcriptional activator [1–4] PRS8 TRIP1 P62195 subunit RPT6, p45 ATPase subunit of PA700, the regulatory P45/SUG1 Proteasome 26S subunit complex of the 26S proteasome [5,6]. ATPase 5. Proteasome subunit p45 Component of the Mediator complex, a Mediator of RNA coactivator involved in the regulated TRIP2 Q15648 MED1 polymerase II transcription of nearly all RNA polymerase II- transcription subunit 1 dependent genes [7]. Thyroid hormone receptor-associated TRAP220 protein complex 220 kDa component Interaction with nuclear receptors in a ligand- Vitamin D receptor- DRIP205 dependent manner, global activator for the interacting protein nuclear receptor superfamily [8–10]. complex component Activator-recruited ARC205 cofactor 205 kDa component Subunit p200 of the transcriptional factor CRSP CRSP200 Cofactor Required for Sp1 is identical to TRIP2 [11]. PBP, PPAR- Peroxisome proliferator- Interaction with peroxisome proliferator- binding activated receptor- activated receptor [12]. protein binding protein Recognized by PAb1801 Identification of common antigenic and RB18A antibody functional properties with P53 [13]. The heterodimer NUFIP1 (nuclear fragile X mental retardation-interacting protein 1)- ZNHI3 Zinc finger HIT ZNHIT3 is implicated in the assembly of the box TRIP3 Q15649 ZNHIT3 domain-containing C/D small nucleolar ribonucleoprotein protein 3 (snoRNP) [14] and in starvation-induced ribophagy [15]. HNF-4a Hepatocyte Nuclear [16] coactivator Factor 4a coactivator Transcription coactivator that associates with Activating signal TRIP4 Q15650 ASC-1 nuclear receptors, transcriptional coactivators cointegrator 1 and basal transcription factors [17]. -

Interactome Analyses Revealed That the U1 Snrnp Machinery Overlaps
www.nature.com/scientificreports OPEN Interactome analyses revealed that the U1 snRNP machinery overlaps extensively with the RNAP II Received: 12 April 2018 Accepted: 24 May 2018 machinery and contains multiple Published: xx xx xxxx ALS/SMA-causative proteins Binkai Chi1, Jeremy D. O’Connell1,2, Tomohiro Yamazaki1, Jaya Gangopadhyay1, Steven P. Gygi1 & Robin Reed1 Mutations in multiple RNA/DNA binding proteins cause Amyotrophic Lateral Sclerosis (ALS). Included among these are the three members of the FET family (FUS, EWSR1 and TAF15) and the structurally similar MATR3. Here, we characterized the interactomes of these four proteins, revealing that they largely have unique interactors, but share in common an association with U1 snRNP. The latter observation led us to analyze the interactome of the U1 snRNP machinery. Surprisingly, this analysis revealed the interactome contains ~220 components, and of these, >200 are shared with the RNA polymerase II (RNAP II) machinery. Among the shared components are multiple ALS and Spinal muscular Atrophy (SMA)-causative proteins and numerous discrete complexes, including the SMN complex, transcription factor complexes, and RNA processing complexes. Together, our data indicate that the RNAP II/U1 snRNP machinery functions in a wide variety of molecular pathways, and these pathways are candidates for playing roles in ALS/SMA pathogenesis. Te neurodegenerative disease Amyotrophic Lateral Sclerosis (ALS) has no known treatment, and elucidation of disease mechanisms is urgently needed. Tis problem has been especially daunting, as mutations in greater than 30 genes are ALS-causative, and these genes function in numerous cellular pathways1. Tese include mitophagy, autophagy, cytoskeletal dynamics, vesicle transport, DNA damage repair, RNA dysfunction, apoptosis, and pro- tein aggregation2–6. -

Gene Ontology Analysis of Arthrogryposis (Multiple Congenital Contractures)
Received: 5 March 2019 Revised: 13 June 2019 Accepted: 17 July 2019 DOI: 10.1002/ajmg.c.31733 RESEARCH ARTICLE Gene ontology analysis of arthrogryposis (multiple congenital contractures) Jeff Kiefer1 | Judith G. Hall2,3 1Systems Oncology, Scottsdale, Arizona Abstract 2Department of Medical Genetics, University of British Columbia and BC Children's In 2016, we published an article applying Gene Ontology Analysis to the genes that had Hospital, Vancouver, British Columbia, Canada been reported to be associated with arthrogryposis (multiple congenital contractures) (Hall 3Department of Pediatrics, University of & Kiefer, 2016). At that time, 320 genes had been reported to have mutations associated British Columbia and BC Children's Hospital, Vancouver, British Columbia, Canada with arthrogryposis. All were associated with decreased fetal movement. These 320 genes were analyzed by biological process and cellular component categories, and yielded 22 Correspondence Judith G. Hall, Department of Medical distinct groupings. Since that time, another 82 additional genes have been reported, now Genetics, BC Children's Hospital, 4500 Oak totaling 402 genes, which when mutated, are associated with arthrogryposis (arthrogryposis Street, Room C234, Vancouver, British Columbia V6H 3N1, Canada. multiplex congenita). So, we decided to update the analysis in order to stimulate further Email: [email protected] research and possible treatment. Now, 29 groupings can be identified, but only 19 groups have more than one gene. KEYWORDS arthrogryposis, developmental pathways, enrichment analysis, gene ontology, multiple congenital contractures 1 | INTRODUCTION polyhydramnios, decreased gut mobility and shortened gut, short umbili- cal cord, skin changes, and multiple joints with limitation of movement, Arthrogryposis is the term that has been used for the last century including limbs, jaw, and spine). -

Using Historical Museum Samples to Examine Divergent and Parallel Evolution in the Invasive 1 Starling 2 Katarina C. Stuart1, W
1 Using historical museum samples to examine divergent and parallel evolution in the invasive 2 starling 3 Katarina C. Stuart1, William B. Sherwin1, Jeremy J. Austin2, Melissa Bateson3, Marcel Eens4, Matthew 4 C. Brandley5,6, Lee A. Rollins1 5 1 Evolution & Ecology Research Centre, School of Biological, Earth and Environmental Sciences, UNSW 6 Sydney, Sydney, New South Wales, Australia 7 2 Australian Centre for Ancient DNA (ACAD), School of Biological Sciences, University of Adelaide, 8 Adelaide, SA, Australia 9 3 Institute of Neuroscience, Newcastle University, Newcastle upon Tyne, UK 10 4 Department of Biology, Behavioural Ecology and Ecophysiology Group, University of Antwerp, 2610 11 Wilrijk, Belgium 12 5 Section of Amphibians and Reptiles, Carnegie Museum of Natural History, Pittsburgh, PA, USA. 13 6 Powdermill Nature Reserve, Carnegie Museum of Natural History, Rector, PA, USA. 14 15 Supplementary Materials 16 Supplementary Material: Appendix 1 17 Alternate variant calling pipelines 18 In addition to the BWA aln pipeline, the BWA mem and GATK variant calling pipeline was run on the 19 cleaned and processed raw data produced by process_radtags. BWA mem was run on default 20 parameters, before being processed by STACKS gstacks and populations. For the GATK pipeline, 21 BOWTIE2 was used for alignment (--phred33 --very-sensitive-local –I), SAMTOOLS to produce a sorted 22 bam file. The PICARD v2.18.26 (under Java v8u121) BuildBamIndex function was used to index the 23 reads. The GATK HaplotypeCaller function was used to call SNPs and assemble the haplotypes 24 separately for eachs ample. The GATK functions CombineGVCFs and GenotypeGVCFs were used to 25 combine each individual gvcf file into one vcf file for all individuals 26 For comparison to the primary variant data set, two filtering parameters were used.