Sequencing Alignment I Outline: Sequence Alignment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Comparative Analysis of Multiple Sequence Alignment Tools
I.J. Information Technology and Computer Science, 2018, 8, 24-30 Published Online August 2018 in MECS (http://www.mecs-press.org/) DOI: 10.5815/ijitcs.2018.08.04 Comparative Analysis of Multiple Sequence Alignment Tools Eman M. Mohamed Faculty of Computers and Information, Menoufia University, Egypt E-mail: [email protected]. Hamdy M. Mousa, Arabi E. keshk Faculty of Computers and Information, Menoufia University, Egypt E-mail: [email protected], [email protected]. Received: 24 April 2018; Accepted: 07 July 2018; Published: 08 August 2018 Abstract—The perfect alignment between three or more global alignment algorithm built-in dynamic sequences of Protein, RNA or DNA is a very difficult programming technique [1]. This algorithm maximizes task in bioinformatics. There are many techniques for the number of amino acid matches and minimizes the alignment multiple sequences. Many techniques number of required gaps to finds globally optimal maximize speed and do not concern with the accuracy of alignment. Local alignments are more useful for aligning the resulting alignment. Likewise, many techniques sub-regions of the sequences, whereas local alignment maximize accuracy and do not concern with the speed. maximizes sub-regions similarity alignment. One of the Reducing memory and execution time requirements and most known of Local alignment is Smith-Waterman increasing the accuracy of multiple sequence alignment algorithm [2]. on large-scale datasets are the vital goal of any technique. The paper introduces the comparative analysis of the Table 1. Pairwise vs. multiple sequence alignment most well-known programs (CLUSTAL-OMEGA, PSA MSA MAFFT, BROBCONS, KALIGN, RETALIGN, and Compare two biological Compare more than two MUSCLE). -

Chapter 6: Multiple Sequence Alignment Learning Objectives
Chapter 6: Multiple Sequence Alignment Learning objectives • Explain the three main stages by which ClustalW performs multiple sequence alignment (MSA); • Describe several alternative programs for MSA (such as MUSCLE, ProbCons, and TCoffee); • Explain how they work, and contrast them with ClustalW; • Explain the significance of performing benchmarking studies and describe several of their basic conclusions for MSA; • Explain the issues surrounding MSA of genomic regions Outline: multiple sequence alignment (MSA) Introduction; definition of MSA; typical uses Five main approaches to multiple sequence alignment Exact approaches Progressive sequence alignment Iterative approaches Consistency-based approaches Structure-based methods Benchmarking studies: approaches, findings, challenges Databases of Multiple Sequence Alignments Pfam: Protein Family Database of Profile HMMs SMART Conserved Domain Database Integrated multiple sequence alignment resources MSA database curation: manual versus automated Multiple sequence alignments of genomic regions UCSC, Galaxy, Ensembl, alignathon Perspective Multiple sequence alignment: definition • a collection of three or more protein (or nucleic acid) sequences that are partially or completely aligned • homologous residues are aligned in columns across the length of the sequences • residues are homologous in an evolutionary sense • residues are homologous in a structural sense Example: 5 alignments of 5 globins Let’s look at a multiple sequence alignment (MSA) of five globins proteins. We’ll use five prominent MSA programs: ClustalW, Praline, MUSCLE (used at HomoloGene), ProbCons, and TCoffee. Each program offers unique strengths. We’ll focus on a histidine (H) residue that has a critical role in binding oxygen in globins, and should be aligned. But often it’s not aligned, and all five programs give different answers. -

How to Generate a Publication-Quality Multiple Sequence Alignment (Thomas Weimbs, University of California Santa Barbara, 11/2012)
Tutorial: How to generate a publication-quality multiple sequence alignment (Thomas Weimbs, University of California Santa Barbara, 11/2012) 1) Get your sequences in FASTA format: • Go to the NCBI website; find your sequences and display them in FASTA format. Each sequence should look like this (http://www.ncbi.nlm.nih.gov/protein/6678177?report=fasta): >gi|6678177|ref|NP_033320.1| syntaxin-4 [Mus musculus] MRDRTHELRQGDNISDDEDEVRVALVVHSGAARLGSPDDEFFQKVQTIRQTMAKLESKVRELEKQQVTIL ATPLPEESMKQGLQNLREEIKQLGREVRAQLKAIEPQKEEADENYNSVNTRMKKTQHGVLSQQFVELINK CNSMQSEYREKNVERIRRQLKITNAGMVSDEELEQMLDSGQSEVFVSNILKDTQVTRQALNEISARHSEI QQLERSIRELHEIFTFLATEVEMQGEMINRIEKNILSSADYVERGQEHVKIALENQKKARKKKVMIAICV SVTVLILAVIIGITITVG 2) In a text editor, paste all your sequences together (in the order that you would like them to appear in the end). It should look like this: >gi|6678177|ref|NP_033320.1| syntaxin-4 [Mus musculus] MRDRTHELRQGDNISDDEDEVRVALVVHSGAARLGSPDDEFFQKVQTIRQTMAKLESKVRELEKQQVTIL ATPLPEESMKQGLQNLREEIKQLGREVRAQLKAIEPQKEEADENYNSVNTRMKKTQHGVLSQQFVELINK CNSMQSEYREKNVERIRRQLKITNAGMVSDEELEQMLDSGQSEVFVSNILKDTQVTRQALNEISARHSEI QQLERSIRELHEIFTFLATEVEMQGEMINRIEKNILSSADYVERGQEHVKIALENQKKARKKKVMIAICV SVTVLILAVIIGITITVG >gi|151554658|gb|AAI47965.1| STX3 protein [Bos taurus] MKDRLEQLKAKQLTQDDDTDEVEIAVDNTAFMDEFFSEIEETRVNIDKISEHVEEAKRLYSVILSAPIPE PKTKDDLEQLTTEIKKRANNVRNKLKSMERHIEEDEVQSSADLRIRKSQHSVLSRKFVEVMTKYNEAQVD FRERSKGRIQRQLEITGKKTTDEELEEMLESGNPAIFTSGIIDSQISKQALSEIEGRHKDIVRLESSIKE LHDMFMDIAMLVENQGEMLDNIELNVMHTVDHVEKAREETKRAVKYQGQARKKLVIIIVIVVVLLGILAL IIGLSVGLK -

"Phylogenetic Analysis of Protein Sequence Data Using The
Phylogenetic Analysis of Protein Sequence UNIT 19.11 Data Using the Randomized Axelerated Maximum Likelihood (RAXML) Program Antonis Rokas1 1Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee ABSTRACT Phylogenetic analysis is the study of evolutionary relationships among molecules, phenotypes, and organisms. In the context of protein sequence data, phylogenetic analysis is one of the cornerstones of comparative sequence analysis and has many applications in the study of protein evolution and function. This unit provides a brief review of the principles of phylogenetic analysis and describes several different standard phylogenetic analyses of protein sequence data using the RAXML (Randomized Axelerated Maximum Likelihood) Program. Curr. Protoc. Mol. Biol. 96:19.11.1-19.11.14. C 2011 by John Wiley & Sons, Inc. Keywords: molecular evolution r bootstrap r multiple sequence alignment r amino acid substitution matrix r evolutionary relationship r systematics INTRODUCTION the baboon-colobus monkey lineage almost Phylogenetic analysis is a standard and es- 25 million years ago, whereas baboons and sential tool in any molecular biologist’s bioin- colobus monkeys diverged less than 15 mil- formatics toolkit that, in the context of pro- lion years ago (Sterner et al., 2006). Clearly, tein sequence analysis, enables us to study degree of sequence similarity does not equate the evolutionary history and change of pro- with degree of evolutionary relationship. teins and their function. Such analysis is es- A typical phylogenetic analysis of protein sential to understanding major evolutionary sequence data involves five distinct steps: (a) questions, such as the origins and history of data collection, (b) inference of homology, (c) macromolecules, developmental mechanisms, sequence alignment, (d) alignment trimming, phenotypes, and life itself. -

Aligning Reads: Tools and Theory Genome Transcriptome Assembly Mapping Mapping
Aligning reads: tools and theory Genome Transcriptome Assembly Mapping Mapping Reads Reads Reads RSEM, STAR, Kallisto, Trinity, HISAT2 Sailfish, Scripture Salmon Splice-aware Transcript mapping Assembly into Genome mapping and quantification transcripts htseq-count, StringTie Trinotate featureCounts Transcript Novel transcript Gene discovery & annotation counting counting Homology-based BLAST2GO Novel transcript annotation Transcriptome Mapping Reads RSEM, Kallisto, Sailfish, Salmon Transcript mapping and quantification Transcriptome Biological samples/Library preparation Mapping Reads RSEM, Kallisto, Sequence reads Sailfish, Salmon FASTQ (+reference transcriptome index) Transcript mapping and quantification Quantify expression Salmon, Kallisto, Sailfish Pseudocounts DGE with R Functional Analysis with R Goal: Finding where in the genome these reads originated from 5 chrX:152139280 152139290 152139300 152139310 152139320 152139330 Reference --->CGCCGTCCCTCAGAATGGAAACCTCGCT TCTCTCTGCCCCACAATGCGCAAGTCAG CD133hi:LM-Mel-34pos Normal:HAH CD133lo:LM-Mel-34neg Normal:HAH CD133lo:LM-Mel-14neg Normal:HAH CD133hi:LM-Mel-34pos Normal:HAH CD133lo:LM-Mel-42neg Normal:HAH CD133hi:LM-Mel-42pos Normal:HAH CD133lo:LM-Mel-34neg Normal:HAH CD133hi:LM-Mel-34pos Normal:HAH CD133lo:LM-Mel-14neg Normal:HAH CD133hi:LM-Mel-14pos Normal:HAH CD133lo:LM-Mel-34neg Normal:HAH CD133hi:LM-Mel-34pos Normal:HAH CD133lo:LM-Mel-42neg DBTSS:human_MCF7 CD133hi:LM-Mel-42pos CD133lo:LM-Mel-14neg CD133lo:LM-Mel-34neg CD133hi:LM-Mel-34pos CD133lo:LM-Mel-42neg CD133hi:LM-Mel-42pos CD133hi:LM-Mel-42poschrX:152139280 -

Alignment of Next-Generation Sequencing Data
Gene Expression Analyses Alignment of Next‐Generation Sequencing Data Nadia Lanman HPC for Life Sciences 2019 What is sequence alignment? • A way of arranging sequences of DNA, RNA, or protein to identify regions of similarity • Similarity may be a consequence of functional, structural, or evolutionary relationships between sequences • In the case of NextGen sequencing, alignment identifies where fragments which were sequenced are derived from (e.g. which gene or transcript) • Two types of alignment: local and global http://www‐personal.umich.edu/~lpt/fgf/fgfrseq.htm Global vs Local Alignment • Global aligners try to align all provided sequence end to end • Local aligners try to find regions of similarity within each provided sequence (match your query with a substring of your subject/target) gap mismatch Alignment Example Raw sequences: A G A T G and G A T TG 2 matches, 0 4 matches, 1 4 matches, 1 3 matches, 2 gaps insertion insertion end gaps A G A T G A G A ‐ T G . A G A T ‐ G . A G A T G . G A T TG . G A T TG . G A T TG . G A T TG NGS read alignment • Allows us to determine where sequence fragments (“reads”) came from • Quantification allows us to address relevant questions • How do samples differ from the reference genome • Which genes or isoforms are differentially expressed Haas et al, 2010, Nature. Standard Differential Expression Analysis Differential Check data Unsupervised expression quality Clustering analysis Trim & filter Count reads GO enrichment reads, remove aligning to analysis adapters each gene Align reads to Check -

Errors in Multiple Sequence Alignment and Phylogenetic Reconstruction
Multiple Sequence Alignment Errors and Phylogenetic Reconstruction THESIS SUBMITTED FOR THE DEGREE “DOCTOR OF PHILOSOPHY” BY Giddy Landan SUBMITTED TO THE SENATE OF TEL-AVIV UNIVERSITY August 2005 This work was carried out under the supervision of Professor Dan Graur Acknowledgments I would like to thank Dan for more than a decade of guidance in the fields of molecular evolution and esoteric arts. This study would not have come to fruition without the help, encouragement and moral support of Tal Dagan and Ron Ophir. To them, my deepest gratitude. Time flies like an arrow Fruit flies like a banana - Groucho Marx Table of Contents Abstract ..........................................................................................................................1 Chapter 1: Introduction................................................................................................5 Sequence evolution...................................................................................................6 Alignment Reconstruction........................................................................................7 Errors in reconstructed MSAs ................................................................................10 Motivation and aims...............................................................................................13 Chapter 2: Methods.....................................................................................................17 Symbols and Acronyms..........................................................................................17 -

Tunca Doğan , Alex Bateman , Maria J. Martin Your Choice
(—THIS SIDEBAR DOES NOT PRINT—) UniProt Domain Architecture Alignment: A New Approach for Protein Similarity QUICK START (cont.) DESIGN GUIDE Search using InterPro Domain Annotation How to change the template color theme This PowerPoint 2007 template produces a 44”x44” You can easily change the color theme of your poster by going to presentation poster. You can use it to create your research 1 1 1 the DESIGN menu, click on COLORS, and choose the color theme of poster and save valuable time placing titles, subtitles, text, Tunca Doğan , Alex Bateman , Maria J. Martin your choice. You can also create your own color theme. and graphics. European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), We provide a series of online tutorials that will guide you Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK through the poster design process and answer your poster Correspondence: [email protected] production questions. To view our template tutorials, go online to PosterPresentations.com and click on HELP DESK. ABSTRACT METHODOLOGY RESULTS & DISCUSSION When you are ready to print your poster, go online to InterPro Domains, DAs and DA Alignment PosterPresentations.com Motivation: Similarity based methods have been widely used in order to Generation of the Domain Architectures: You can also manually change the color of your background by going to VIEW > SLIDE MASTER. After you finish working on the master be infer the properties of genes and gene products containing little or no 1) Collect the hits for each protein from InterPro. Domain annotation coverage Overlap domain hits problem in Need assistance? Call us at 1.510.649.3001 difference b/w domain databases: the InterPro database: sure to go to VIEW > NORMAL to continue working on your poster. -

Developing and Implementing an Institute-Wide Data Sharing Policy Stephanie OM Dyke and Tim JP Hubbard*
Dyke and Hubbard Genome Medicine 2011, 3:60 http://genomemedicine.com/content/3/9/60 CORRESPONDENCE Developing and implementing an institute-wide data sharing policy Stephanie OM Dyke and Tim JP Hubbard* Abstract HapMap Project [7], also decided to follow HGP prac- tices and to share data publicly as a resource for the The Wellcome Trust Sanger Institute has a strong research community before academic publications des- reputation for prepublication data sharing as a result crib ing analyses of the data sets had been prepared of its policy of rapid release of genome sequence (referred to as prepublication data sharing). data and particularly through its contribution to the Following the success of the first phase of the HGP [8] Human Genome Project. The practicalities of broad and of these other projects, the principles of rapid data data sharing remain largely uncharted, especially to release were reaffirmed and endorsed more widely at a cover the wide range of data types currently produced meeting of genomics funders, scientists, public archives by genomic studies and to adequately address and publishers in Fort Lauderdale in 2003 [9]. Meanwhile, ethical issues. This paper describes the processes the Organisation for Economic Co-operation and and challenges involved in implementing a data Develop ment (OECD) Committee on Scientific and sharing policy on an institute-wide scale. This includes Tech nology Policy had established a working group on questions of governance, practical aspects of applying issues of access to research information [10,11], which principles to diverse experimental contexts, building led to a Declaration on access to research data from enabling systems and infrastructure, incentives and public funding [12], and later to a set of OECD guidelines collaborative issues. -

Molecular Homology and Multiple-Sequence Alignment: an Analysis of Concepts and Practice
CSIRO PUBLISHING Australian Systematic Botany, 2015, 28, 46–62 LAS Johnson Review http://dx.doi.org/10.1071/SB15001 Molecular homology and multiple-sequence alignment: an analysis of concepts and practice David A. Morrison A,D, Matthew J. Morgan B and Scot A. Kelchner C ASystematic Biology, Uppsala University, Norbyvägen 18D, Uppsala 75236, Sweden. BCSIRO Ecosystem Sciences, GPO Box 1700, Canberra, ACT 2601, Australia. CDepartment of Biology, Utah State University, 5305 Old Main Hill, Logan, UT 84322-5305, USA. DCorresponding author. Email: [email protected] Abstract. Sequence alignment is just as much a part of phylogenetics as is tree building, although it is often viewed solely as a necessary tool to construct trees. However, alignment for the purpose of phylogenetic inference is primarily about homology, as it is the procedure that expresses homology relationships among the characters, rather than the historical relationships of the taxa. Molecular homology is rather vaguely defined and understood, despite its importance in the molecular age. Indeed, homology has rarely been evaluated with respect to nucleotide sequence alignments, in spite of the fact that nucleotides are the only data that directly represent genotype. All other molecular data represent phenotype, just as do morphology and anatomy. Thus, efforts to improve sequence alignment for phylogenetic purposes should involve a more refined use of the homology concept at a molecular level. To this end, we present examples of molecular-data levels at which homology might be considered, and arrange them in a hierarchy. The concept that we propose has many levels, which link directly to the developmental and morphological components of homology. -

The Uniprot Knowledgebase BLAST
Introduction to bioinformatics The UniProt Knowledgebase BLAST UniProtKB Basic Local Alignment Search Tool A CRITICAL GUIDE 1 Version: 1 August 2018 A Critical Guide to BLAST BLAST Overview This Critical Guide provides an overview of the BLAST similarity search tool, Briefly examining the underlying algorithm and its rise to popularity. Several WeB-based and stand-alone implementations are reviewed, and key features of typical search results are discussed. Teaching Goals & Learning Outcomes This Guide introduces concepts and theories emBodied in the sequence database search tool, BLAST, and examines features of search outputs important for understanding and interpreting BLAST results. On reading this Guide, you will Be aBle to: • search a variety of Web-based sequence databases with different query sequences, and alter search parameters; • explain a range of typical search parameters, and the likely impacts on search outputs of changing them; • analyse the information conveyed in search outputs and infer the significance of reported matches; • examine and investigate the annotations of reported matches, and their provenance; and • compare the outputs of different BLAST implementations and evaluate the implications of any differences. finding short words – k-tuples – common to the sequences Being 1 Introduction compared, and using heuristics to join those closest to each other, including the short mis-matched regions Between them. BLAST4 was the second major example of this type of algorithm, From the advent of the first molecular sequence repositories in and rapidly exceeded the popularity of FastA, owing to its efficiency the 1980s, tools for searching dataBases Became essential. DataBase searching is essentially a ‘pairwise alignment’ proBlem, in which the and Built-in statistics. -
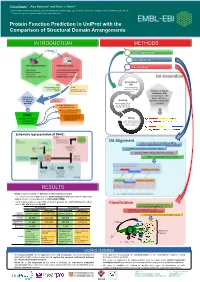
Protein Function Prediction in Uniprot with the Comparison of Structural Domain Arrangements
Tunca Dogan1,*, Alex Bateman1 and Maria J. Martin1 1 European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK * To whom correspondence should be addressed: [email protected] Protein Function Prediction in UniProt with the Comparison of Structural Domain Arrangements INTRODUCTION METHODS Data Preparation DA alignment Classification DA Generation Generation of SAAS DAs (Automatic decision tree- UniRule using InterProScan based rule-generating (Manually curated rules results on UniProtKB system) created by curation team) proteins Definition of domain architecture (DA): concatenation of the Requirement of InterPro IDs of the new approaches Annotations from Grouping for automatic other sources domains on the protein annotation proteins under shared sequence Treats domain hits as DAs & separation of strings (instead of a.a.) learning and test sets Pairwise alignment of DAs for similarity detection DAAC Supervised classification (Domain Architecture of query sequences into Mining functional classes Alignment and functional annotations Classification) from source databases (for learning set) Schematic representation of DAAC: DA Alignment A modified version of Needleman-Wunsch global sequence alignment algorithm to compare the DAs by: - treating the domains as the strings in a sequence - working for 7497 InterPro domains instead of 20 a.a - fast due to reduced total number of operations RESULTS • DAAC system is trained for GO term and EC number prediction