Postmortem Examination of Member III-13
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Dual RNA-Seq Analysis of Mus Musculus and Leishmania Donovani Transcriptomes
Dual RNA-Seq analysis of Mus musculus and Leishmania donovani transcriptomes ALEXANDRA HART MASTER OF SCIENCE BY RESEARCH UNIVERSITY OF YORK BIOLOGY DECEMBER 2017 ABSTRACT Parasitic protozoa of the genus Leishmania cause a spectrum of disease, affecting 12 million people worldwide. This project aimed to investigate the effect of Leishmania donovani infection on the gene expression of healthy/WT (Black 6) and immunocompromised (RAG2KO) mice. Differences in the gene expression of parasites in inoculum and tissue were also elucidated. WT and RAG mice were infected using an L. donovani inoculum, and were euthanised after 28 days. Harvested spleens (and the inoculum) were used to generate RNA samples, from which mRNA was isolated and purified. Transcriptome data was generated using dual RNA-Seq approaches from the mRNA samples. After appropriate pre-processing, data underwent a number of bioinformatic analyses to explore differential gene expression, such as Gene Ontology, Gene Set Enrichment, and KEGG Pathway analysis. Comparison of different mouse spleen transcriptomes revealed that even in uninfected mice, WT mice more highly express genes related to immunoglobulins when compared with their immunocompromised counterparts. Healthy mice were found to react to infection through the induction of inflammatory response, and the production of NOX generating species. RAG mice still upregulated immunoglobulin-related genes in response to infection, despite their inability to generate antibodies, T-cells, and B-cells. However, RAG modulation of haeme and iron metabolism may contribute to defence against the parasites despite a lack of acquired immunity. Differences in the amastin, the key glycoprotein on the surface on intracellular-stage parasites, are apparent between the inoculum and tissue parasites, which may reflect microenvironment adaptation. -

Facteur De Risque Génétique Aux Maladies Inflammatoires De L’Intestin Et Modulateur D’Inflammation
Université de Montréal MAST3 : facteur de risque génétique aux maladies inflammatoires de l’intestin et modulateur d’inflammation par Catherine Labbé Département de sciences biomédicales Faculté de médecine Thèse présentée à la Faculté de médecine en vue de l’obtention du grade de doctorat en sciences biomédicales 5 août, 2011 © Catherine Labbé, 2011 Université de Montréal Faculté de médecine Cette thèse intitulée : MAST3 : facteur de risque génétique aux maladies inflammatoires de l’intestin et modulateur d’inflammation Présentée par : Catherine Labbé a été évaluée par un jury composé des personnes suivantes : Daniel Sinnett, président-rapporteur John D. Rioux, directeur de recherche Zoha Kibar, membre du jury Yohan Bossé, examinateur externe Gaëtan Mayer, représentant du doyen de la FES i Résumé La maladie de Crohn (MC) et la colite ulcéreuse (CU) sont des maladies inflammatoires chroniques du tube digestif qu’on regroupe sous le terme maladies inflammatoires de l’intestin (MII). Les mécanismes moléculaires menant au développement des MII ne sont pas entièrement connus, mais des études génétiques et fonctionnelles ont permis de mettre en évidence des interactions entre des prédispositions génétiques et des facteurs environnementaux - notamment la flore intestinale – qui contribuent au développement d’une dérégulation de la réponse immunitaire menant à l’inflammation de la muqueuse intestinale. Des études d’association pangénomiques et ciblées ont permis d’identifier plusieurs gènes de susceptibilité aux MII mais les estimations de la contribution de ces gènes à l’héritabilité suggèrent que plusieurs gènes restent à découvrir. Certains d’entre eux peuvent se trouver dans les régions identifiées par des études de liaison génétique. -
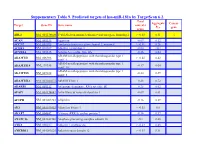
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Characterizing Alternative Splicing and Long Non
CHARACTERIZING ALTERNATIVE SPLICING AND LONG NON-CODING RNA WITH HIGH-THROUGHPUT SEQUENCING TECHNOLOGY Ao Zhou Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the School of Informatics and Computing, Indiana University October 2018 Accepted by the Graduate Faculty of Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Doctoral Committee ______________________________________ Huanmei Wu, Ph.D., Chair ______________________________________ Yunlong Liu, Ph.D. October 2018 ______________________________________ Sarath C. Janga, Ph.D. ______________________________________ Xiaowen Liu, Ph.D. ii © 2018 Ao Zhou iii DEDICATION To my parents, who grew the love of science in me and constantly supported me from the beginning of my life. iv ACKNOWLEDGEMENTS The 10 years’ odyssey of Ph.D. study was full of ups and downs. During the past decade, I worked with many people, I learned from many people, I received generous help from many people, and I shared happiness with many people. First of all, I would like to thank Prof. Yunlong Liu, who led me in the gate of next generation sequencing technology, and continuously helped me to improve my scientific knowledge and skills during the past 8 years. Besides his selfless support in academics, his optimism and “can do” spirit inspired me during the down times, and will continue inspiring me in my future career. I would also like to thank Prof. Jake Chen, who taught me a lot on database construction and management skills. He tremendously influenced me with his scientific way of thinking and logical presentations skills.