CU-Catalyzed Enantioselective Conjugate Addition of Organometal Reagents to Unsaturated Carbonyls : an Enantioselective Total Synthesis of Clavirolide C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent (19) 11 Patent Number: 5,760,032 Kitajima Et Al
USOO576.0032A United States Patent (19) 11 Patent Number: 5,760,032 Kitajima et al. 45) Date of Patent: Jun. 2, 1998 54) THIENYLAZOLE COMPOUND AND OTHER PUBLICATIONS THENOTRAZOLODIAZEPINE CO UND T. Tahara et al. "Syntheses and Structure-Activity Relation 75) Inventors: Hiroshi Kitajima; Syuji Ehara; ships of 6-Aryl-4H-s-triazolo3.4-cithieno23-e 14-di Hideaki Sato, all of Fukuoka; Minoru azepines" Arzneimittel Forschung Drug Research, vol. 28. Moriwaki, Osaka; Kenichi Onishi, 1978. Fukuoka, all of Japan Waiser, et al. J. Med. Chem. (1991). 34(3), 1209-21. G.N. Woodruff et al., "Cholecyst Antagonists". Annu. Rev. 73) Assignee: Yoshitomi Pharmaceutical Industries, Pharmacol. Toxicol., vol. 31, pp. 469-501, 1991. Ltd., Osaka, Japan J. Martinez et al., "Synthesis and Biological Activity of New Peptide Segments of Gastrin Exhibiting Gastrin Antagonist 21 Appl. No.: 750,025 Property". J. Med. Chem. vol. 27. pp. 1597-1601, 1984. 1. B.E. Evans, "Methods for Drug Discovery: Development of 22 PCT Filed: Jun. 1, 1995 Potent, Selective. Orally Effective Cholecystokinin Antago 86 PCT No.: PCT/JP95/01071 nists". J. Med. Chem., vol. 31, pp. 2235-2246, 1988. S371 Date: Nov. 22, 1996 Primary Examiner Mukund J. Shah Assistant Examiner-Bruck Kifle S 102(e) Date: Nov. 22, 1996 Attorney; Agent, or Firn-Wenderoth. Lind & Ponack 87 PCT Pub. No.: WO95/32964 57 ABSTRACT CT Pub. Date: Dec. 7, 1995 Thienylazole compounds (I) and thienotriazolodiazepine 30 Foreign Application Priority Data compounds (II) of the formulas Jun. 1, 1994 (JP) Japan ...................................... 6-00889 (I) (51) Int. Cl. ..................... A61K 31/55; CO7D 491/00; C07D 513/00; CO7D 515/00 52 U.S. -

Sigma Biochemical Condensed Phase
Sigma Biochemical Condensed Phase Library Listing – 10,411 spectra This library provides a comprehensive spectral collection of the most common chemicals found in the Sigma Biochemicals and Reagents catalog. It includes an extensive combination of spectra of interest to the biochemical field. The Sigma Biochemical Condensed Phase Library contains 10,411 spectra acquired by Sigma-Aldrich Co. which were examined and processed at Thermo Fisher Scientific. These spectra represent a wide range of chemical classes of particular interest to those engaged in biochemical research or QC. The spectra include compound name, molecular formula, CAS (Chemical Abstract Service) registry number, and Sigma catalog number. Sigma Biochemical Condensed Phase Index Compound Name Index Compound Name 8951 (+)-1,2-O-Isopropylidene-sn-glycerol 4674 (+/-)-Epinephrine methyl ether .HCl 7703 (+)-10-Camphorsulfonic acid 8718 (+/-)-Homocitric acid lactone 10051 (+)-2,2,2-Trifluoro-1-(9-anthryl)ethanol 4739 (+/-)-Isoproterenol .HCl 8016 (+)-2,3-Dibenzoyl-D-tartaric acid 4738 (+/-)-Isoproterenol, hemisulfate salt 8948 (+)-2,3-O-Isopropylidene-2,3- 5031 (+/-)-Methadone .HCl dihydroxy-1,4-bis- 9267 (+/-)-Methylsuccinic acid (diphenylphosphino)but 9297 (+/-)-Miconazole, nitrate salt 6164 (+)-2-Octanol 9361 (+/-)-Nipecotic acid 9110 (+)-6-Methoxy-a-methyl-2- 9618 (+/-)-Phenylpropanolamine .HCl naphthaleneacetic acid 4923 (+/-)-Sulfinpyrazone 7271 (+)-Amethopterin 10404 (+/-)-Taxifolin 4368 (+)-Bicuculline 4469 (+/-)-Tetrahydropapaveroline .HBr 7697 (+)-Camphor 4992 (+/-)-Verapamil, -

Technical Report for the Substantial Modification Rule for 62-730, F.A.C
Technical Report for the Substantial Modification Rule for 62-730, F.A.C. Prepared for the Hazardous Waste Regulation Section Florida Department of Environmental Protection By Kendra F. Goff, Ph. D. Stephen M. Roberts, Ph.D. Center for Environmental & Human Toxicology University of Florida Gainesville, Florida August 1, 2008 During an adverse event, such as a fire or explosion, hazardous materials from a storage or transfer facility may be released into the atmosphere. The potential human health impacts of such a release are assessed through air modeling coupled with chemical-specific inhalation criteria for short-term exposures. With this approach, distances from the facility over which differing levels of health effects from inhalation of airborne hazardous materials are expected can be predicted. Because the movement in air and toxic potency varies among chemicals, the potential impact area for a facility is dependent upon the chemicals present and their quantities. The impact area can be calculated for a facility under current and possible future conditions. This information can be used to determine whether proposed changes in storage conditions or the nature and quantities of chemicals handled would result in a substantial difference in the area of potential impact. The following sections provide technical guidance on how impact areas for facilities handling hazardous materials under Chapter 62-730, F.A.C. should be determined. Air modeling. Any model used for the purposes of demonstration must be capable of producing results in accordance with worst-case scenario provisions of Program 3 of the Accidental Release Prevention Program of s. 112(r)(7) of the Clean Air Act. -

Ep 0776892 A1
Europaisches Patentamt (19) European Patent Office Office europeen des brevets (11) EP 0 776 892 A1 (12) EUROPEAN PATENT APPLICATION published in accordance with Art. 158(3) EPC (43) Date of publication: (51) int. CI 6: C07D 409/04, C07D 409/14, 04.06.1997 Bulletin 1997/23 C07D 495/14, C07D 495/22, (21) Application number: 95920233.4 A61K 31/41, A61K 31/415, A61K 31/55 (22) Date of filing : 01 .06.1 995 (86) International application number: PCT/JP95/01071 (87) International publication number: WO 95/32964 (07.12.1995 Gazette 1995/52) (84) Designated Contracting States: • SATO, Hideaki, AT BE CH DE DK ES FR GB GR IE IT LI LU MC NL Yoshitomi Pharmaceutical Ind. Ltd. PTSE Chikujo-gun Fukuoka 871 (JP) • MORIWAKI, Minoru (30) Priority: 01.06.1994 WO PCT/JP94/00889 Yoshitomi Pharmaceutical Ind. Ltd. Osaka-shi Osaka 541 (JP) (71) Applicant: YOSHITOMI PHARMACEUTICAL • ONISHI, Kenichi INDUSTRIES, LTD. Yoshitomi Pharmaceutical Ind. Ltd. Osaka-shi Osaka 541 (JP) Yoshitomimachi Chikujo-gun Fukuoka 871 (JP) (72) Inventors: (74) Representative: von Kreisler, Alek, Dipl.-Chem. et • KITAJIMA, Hiroshi, al Yoshitomi Pharmac. Ind. Ltd. Patentanwalte, Yoshitomachi, Chikujo-gun Fukuoka 871 (JP) von Kreisler-Selting-Werner, • EHARA, Syuji, Bahnhofsvorplatz 1 (Deichmannhaus) Yoshitomi Pharmaceutical Ind. Ltd., 50667 Koln (DE) Chikujo-gun Fukuoka 871 (JP) (54) THIENYLAZOLE COMPOUND AND THIENOTRIAZOLODIAZEPINE COMPOUND (57) Thienylazole compounds (I) and thienotriazolodiazepine compounds (II) of the formulas < CM <7> CO CO r»- o Q_ LU Printed by Rank Xerox (UK) Business Services 2.14.7/3.4 (Cont. next page) EP 0 776 892 A1 (I) (II) (CH2)n-CH-Y-Z2 I R3 wherein R1 and R2 are hydrogen, halogen, C1-C5 alkyl and the like; -A=B-is -N=N- and the like; R3 and R19 are hydro- gen, CrC5 alkyl and the like; Y is -NHCO, -NHCONH-, -NHCOO- and the like; Z1 and Z2 are aryl, heteroaryl and the like; Ar is halogen-substituted phenyl and the like; and m is 0 or an integer of 1-5. -

Information to Users
INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print hleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back of the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6" x 9" black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. University Microfilms International A Bell & Howell Information Company 300 North Zeeb Road. Ann Arbor. Ml 48106-1346 USA 313/761-4700 800/521-0600 Order Number 9411918 Part 1. Synthesis of arylalkylguanidines as dopamine agonists. Part 2, Section A. Modifications of trimetoquinol and the effects on /3-adrenergic and thromboxane2 receptor A systems. -
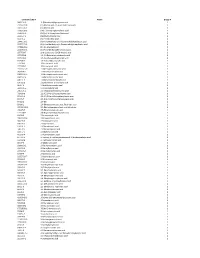
Chemical Classification List
Identifier/CAS # Name Group # 5683-31-8 3-(Trimethylsilyl)propynoic acid 1 21293-29-8 (+)-Abscisic acid, (2, 4-pentadienoic acid) 1 14375-45-2 (±)-Abscisic acid 1 35963-20-3 (1R)-(-)-10-Camphorsulfonic acid 1 3144-16-9 (1S)-(+)-10-Camphorsulfonic acid 1 2444-37-3 (Methylthio)acetic acid 1 611-71-2 (R)-(−)-Mandelic acid 1 20445-31-2 (R)-(+)-α-Methoxy-α-trifluoromethylphenylacetic acid 1 17257-71-5 (S)-(−)-α-Methoxy-α-(trifluoromethyl)phenylacetic acid 1 17199-29-0 (S )-(+)-Mandelic acid 1 26164-26-1 (S)-(+)-α-Methoxyphenylacetic acid 1 1077-28-7 ±)-α-Lipoic acid, DL-6,8-thioctic acid 1 1703-58-8 1,2,3,4-Butanetetracarboxylic acid 1 3971-31-1 1,3-Cyclohexanedicarboxylic acid 1 542-05-2 1,3-Acetonedicarboxylic acid 1 112-38-9 10-Undecenoic acid 1 2777-65-3 10-Undecynoic acid 1 71310-21-9 11-Mercaptoundecanoic acid 1 2834-05-1 11-Bromoundecanoic acid 1 69839-68-5 16-Mercaptohexadecanoic acid 1 4942-47-6 1-Adamantaneacetic acid 1 828-51-3 1-Adamantanecarboxylic acid 1 636-82-8 1-Cyclohexene-1-carboxylic acid 1 86-87-3 1-Naphthaleneacetic acid 1 3443-45-6 1-Pyrenebutyric acid 1 2062-25-1 2-(Trifluoromethyl)cinnamic acid 1 719-60-8 2,3,4,5,6-Pentafluorocinnamic acid 1 653-21-4 2,3,4,5,6-Pentafluorophenylacetic acid 1 94-75-7 2,4-D (2,4-Dichlorophenoxyacetic acid) 1 94-82-6 2,4-DB 1 89-86-1 2,4-Dihydroxybenzoic acid, Resorcylic acid 1 207234-00-2 2,4-Dinitrobenzenesulfonic acid dihydrate 1 490-79-9 2,5-Dihydroxybenzoic acid 1 1141-38-4 2,6-Naphthalenedicarboxylic acid 1 80-58-0 2-Bromobutyric acid 1 70610-87-6 2-Bromooctanoic acid 1 584-93-0 -

Rev. 23 Table 2: Pacs by Chemical Name (Pdf)
Table 2: Protective Action Criteria (PAC) Rev 23 based on applicable AEGLs, ERPGs, or TEELs (chemicals listed in alphabetical order) PAC Rev 23 – August 2007 Table 2 presents an alphabetical listing of the chemicals in the PAC data set and provides Chemical Abstracts Service Registry Numbers (CAS RNs)1, PAC data, and technical comments focusing on changes in the PACs since the previous version of the PAC/TEEL data set (Rev 21). PACs are provided for TEEL-0 (i.e., PAC-0), PAC-1, PAC-2, and PAC-3. Values are usually given in parts per million (ppm) for gases and volatile liquids and in milligrams per cubic meter (mg/m3) for particulate materials (aerosols) and nonvolatile liquids. The columns presented in Table 2 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical The common name of the chemical compound. Compound CAS RN The Chemical Abstracts Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no appreciable risk of health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-3 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Units The units for the PAC values (ppm or mg/m3). Comments Technical comments by the developers of the PAC data set.