The Estimation of Kinetic Parameters in Systems Biology by Comparing Molecular Interaction Fields of Enzymes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

Catalase: Bioinformatics Analyses of One of the Key Enzymes in Hydrogen Peroxide Metabolism
6–71RYHPEHU 2019, Brno, Czech Republic Catalase: Bioinformatics analyses of one of the key enzymes in hydrogen peroxide metabolism Michaela Kameniarova, Romana Kopecka Department of Molecular Biology and Radiobiology Mendel University in Brno Zemedelska 1, 613 00 Brno CZECH REPUBLIC [email protected] Abstract: Catalases (CAT) are family of important antioxidant enzymes present in different isoforms and responsible for scavenging of hydrogen peroxide in almost all aerobically living organisms. In plants, they were found both in unicellular and multicellular species. Here, we performed bioinformatics analysis and analysed catalase evolutionary relationship and expression patterns. By comparing expression profiles of CATs and expression profiles of genes related to abiotic stimuli we found that almost 50% of light signalling genes were co-expressed with CATs. Further, by datamining in available resources and structural modelling we pinpointed candidate amino acid residues responsible for CAT thermostability. Key Words: catalase, H2O2, phylogenetics, abiotic stress, stability INTRODUCTION Plants contain several types of enzymes involved in H2O2 metabolism. These include catalases, ascorbate peroxidases, peroxiredoxins, glutathione/thioredoxin peroxidases, and glutathione S- transferases, but only catalases do not require additional cellular reductants (Mhamdi et al. 2010). Catalases are present almost in all aerobically respiring organisms. Within the cell environment, catalases are localized in all major sites of H2O2 production, mostly at peroxisomes, but they were detected in cytosol, mitochondria, and chloroplasts as well (Sharma and Ahmad 2014). Catalases (CAT, 1.11.1.6) are antioxidant enzymes that catalyse decomposition of hydrogen peroxide to water and molecular oxygen (2H2O2 → 2H2O + O2), an important process in defending cells against oxidative damage caused by reactive oxygen species (ROS; Alfonso-Prieto et al. -

BRENDA Tutorial
BRENDA Tutorial Introduction to the Enzyme Information System Facts about BRENDA (BRaunschweig ENzyme DAtabase) • one of the most comprehensive enzyme information repositories • BRENDA is a member of de.NBI (German Network for Bioinformatics Structure, since 2015) • BRENDA is an ELIXIR Core Data Resource (since 2018) • all enzymes, classified by the Enzyme Nomenclature (IUBMB) • data of molecular biology, biochemistry, medical research, and biotechnology • furthermore BRENDA includes data from interconnected databases containing results from text mining methods and bioinformatic approaches • BRENDA is freely available to the scientific community • more than 80,000 visits of the BRENDA website each month • major updates of the data in BRENDA are performed twice a year History and major developments of BRENDA • BRENDA was created at the former German National Research Center for Biotechnology (GBF, now HZI, Helmholtz Zentrum für Infektionsforschung, Braunschweig, Germany) in 1987 • BRENDA was originally published as a series of book o 1st Edition 1990-1997 (Enzyme Handbook) o 2nd Edition 2001-2013 (Handbook of Enzymes) • BRENDA moved to the University of Cologne, Germany • First online version in 1998 via the SRS system at the EBI • First website of BRENDA in Cologne • Transfer of BRENDA into a fully relational database system • BRENDA moved back to Braunschweig in 2007 • BRENDA is now maintained and further developed at the BRICS - TU Braunschweig Facts about BRENDA The main categories are based on the Enzymes and the Metabolites / Ligands Enzyme-related data encompasses information on: • Enzyme and ligand nomenclature • Organism • Reaction and specificity • Kinetic properties • Structure and role of the ligands • Stability information • Ligand-enzyme information • Enzyme sequence and structure • Mutants and disease • Occurrence, isolation, and preparation • Pathways BRENDA is the most comprehensive information system on: • 7862 EC Numbers (July 2019) • more than 2 Mill. -

Biological Role of Conceptus Derived Factors During Early Pregnancy In
Biological Role of Conceptus Derived Factors During Early Pregnancy in Ruminants A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY IN ANIMAL SCIENCES UNIVERSITY OF MISSOURI- COLUMBIA Division of Animal Science By KELSEY BROOKS Dr. Thomas Spencer, Dissertation Supervisor August 2016 The undersigned have examined the dissertation entitled, BIOLOGICAL ROLE OF CONCEPTUS DERIVED FACTORS DURING EARLY PREGNANCY IN RUMINANTS presented by Kelsey Brooks, a candidate for the degree of doctor of philosophy, and hereby certify that, in their opinion, it is worthy of acceptance. __________________________________ Chair, Dr. Thomas Spencer ___________________________________ Dr. Rodney Geisert ___________________________________ Dr. Randall Prather ___________________________________ Dr. Laura Schulz ACKNOWLEDGMENTS I would like to acknowledge all the students, faculty and staff at Washington State University and the University of Missouri for their help and support throughout my doctoral program. I am grateful for the opportunity to work with Dr. Thomas Spencer, and thank him for his input and guidance not only in planning experiments and completing projects but for helping me turn my love of science into a career in research. I would also like to acknowledge the members of my graduate committee at Washington State University for their help and input during the first 3 years of my studies. A special thanks to Dr. Jim Pru and Cindy Pru for providing unlimited entertainment, and the occasional missing reagent. Thank you to my committee members at the University of Missouri for adopting me late in my program and helping shape my future as an independent scientist. Thanks are also extended to members of the Prather lab and Wells lab for letting me in on the secrets of success using the CRISPR/Cas9 system. -

Investigating the Role of ADP-Forming Acetyl-Coa Synthetase from The
Clemson University TigerPrints All Dissertations Dissertations 5-2016 Investigating the Role of ADP-forming Acetyl-CoA Synthetase from the Protozoan Parasite Entamoeba histolytica Cheryl Page Jones Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/all_dissertations Recommended Citation Jones, Cheryl Page, "Investigating the Role of ADP-forming Acetyl-CoA Synthetase from the Protozoan Parasite Entamoeba histolytica" (2016). All Dissertations. 1668. https://tigerprints.clemson.edu/all_dissertations/1668 This Dissertation is brought to you for free and open access by the Dissertations at TigerPrints. It has been accepted for inclusion in All Dissertations by an authorized administrator of TigerPrints. For more information, please contact [email protected]. INVESTIGATING THE ROLE OF ADP-FORMING ACETYL-COA SYNTHETASE FROM THE PROTOZOAN PARASITE ENTAMOEBA HISTOLYTICA A Dissertation Presented to the Graduate School of Clemson University In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Biochemistry and Molecular Biology by Cheryl Page Jones May 2016 Accepted by Dr. Cheryl Ingram-Smith, Committee Chair Dr. William Marcotte, Jr. Dr. Julia Frugoli Dr. Kimberly Paul ABSTRACT ADP-forming acetyl-CoA synthetase (ACD; EC 6.2.1.13) catalyzes the reversible conversion of acetyl-CoA to acetate coupled to the production of ATP. This enzyme is present only in certain acetate-producing archaea and a limited number of bacteria and eukaryotes. ACD belongs to the same NDP-forming acyl-CoA synthetase enzyme superfamily as succinyl-CoA synthetase (SCS; EC 6.2.1.4) from the citric acid cycle, and a similar three-step mechanism involving a phosphoenzyme intermediate was originally proposed for this enzyme. -
Enzymes and Protein Structure Last Week… PTM’S We (Re)Learned About
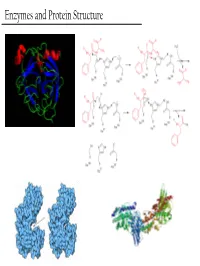
Enzymes and Protein Structure Last Week… PTM’s We (Re)Learned About Primary Structure And Tertiary Structure S-Q-D-A-G-M-Q-Q-G-A-D-M-D-Q-V-S-A Secondary Structure Enzymes What are these crazy things called ‘Enzymes’? And… Why do we care about them? - Enzymes are the catalysts of biology. TS They solve this problem This reaction will go, but it’ll take a G gazillion years… + OH- or You could heat it up to a gazillion degrees RC Enzymes Allow… - Higher reaction rates (same as any catalyst) Catalase - The rates of Enzyme reactions are 106 – 2H2O2 → 2H2O + O2 1012 times those of the corresponding Pretty darn fast: uncatalized reaction… ~ 6 * 105 sec-1 - The rates of reaction for the fastest enzymes are diffusion limited, around 106 turnovers per second. Enzymes Allow… - Milder reaction conditions (same as any catalyst, but better) - We won’t have to heat the reaction up to a gazillion degrees to make it go at a reasonable pace β-strand 4 β-strand 1 α-helix 2 β-strand 3 β-strand 2 - Enzymes from hythermophiles work well at around 85°C, but not at room temp!! β-strand 5 α-helix 1 Acylphosphatase from Sulfolobus solfataricus Enzymes Allow… - Reaction Specificity (better than inorganic catalysts) - Can’t really do this with an inorganic catalyst. A set of similar molecules will likely all be reactive… Enzymes Allow… - Capacity for Regulation (way better than inorganic catalysts) - For inorganic catalysts, you can really only interfere with their activity by using competitive inhibitors or permanent covalent deactivation E.g. -

Clinical Study Normal Intelligence in Female and Male Patients with Congenital Adrenal Hyperplasia
Hindawi Publishing Corporation International Journal of Pediatric Endocrinology Volume 2010, Article ID 853103, 6 pages doi:10.1155/2010/853103 Clinical Study Normal Intelligence in Female and Male Patients with Congenital Adrenal Hyperplasia Sheri A. Berenbaum,1 Kristina Korman Bryk,2 and Stephen C. Duck3 1 Departments of Psychology and Pediatrics, The Pennsylvania State University, University Park, PA 16803, USA 2 Department of Psychology, The Pennsylvania State University, University Park, PA 16803, USA 3 Department of Pediatrics, Pritzker School of Medicine, University of Chicago, and Pediatric Endocrinology, North Shore University Health Services, 9977 Woods Drive, Skokie, IL 60077, USA Correspondence should be addressed to Sheri A. Berenbaum, [email protected] Received 25 May 2010; Revised 30 July 2010; Accepted 16 August 2010 Academic Editor: Todd Nebesio Copyright © 2010 Sheri A. Berenbaum et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. We provide evidence regarding the nature, causes, and consequences of intelligence in patients with 21-hydroxylase deficient congenital adrenal hyperplasia (CAH). Intelligence and quality of life (psychological adjustment) were measured on multiple occasions from childhood to young adulthood in 104 patients with CAH (62 females, 42 males) and 88 unaffected relatives (31 females, 57 males). Information on disease severity (CAH type, age at diagnosis, genital virilization for girls) and salt-wasting crises was obtained from medical records. There was no evidence of intellectual deficit in either female or male patients with CAH. Intelligence was not significantly associated with psychological adjustment or disease characteristics. -

VU Research Portal
VU Research Portal Challenging metabolic networks at high temperature: Kouril, T. 2012 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Kouril, T. (2012). Challenging metabolic networks at high temperature: The central carbohydrate metabolism of Sulfolobus solfataricus. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 23. Sep. 2021 Chapter 4 DISCUSSION DISCUSSION Chapter 4 DISCUSSION Over the past years, “extremophiles” and especially (hyper)thermophiles have received much attention because they are discussed as the closest relatives to the last universal common ancestor of life on earth (Forterre, 1996). (Hyper)thermophilic Archaea function as model organisms to study adaptation strategies to high temperature. Furthermore, the application of enzymes from (hyper)thermophiles as well as (hyper)thermophilic production strains are considered to have a high potential for many industrial, molecular biology, and biotechnological applications (Van den Burg, 2003). -

Full Text (PDF)
Pathophysiology/Complications ORIGINAL ARTICLE Colesevelam Improves Oral but Not Intravenous Glucose Tolerance by a Mechanism Independent of Insulin Sensitivity and b-Cell Function ANNA L. MARINA, MD BRENDA K. MONTGOMERY, RN, MS, CDE currently under investigation with studies KRISTINA M. UTZSCHNEIDER, MD SANTICA M. MARCOVINA, PHD, SCD reporting conflicting results (6). There is a LORENA A. WRIGHT, MD STEVEN E. KAHN, MB, CHB complex interaction between FXR and LXR, which often have counterbalancing effects (7). LXR has been described as a OBJECTIVEd To determine the mechanism by which the bile acid sequestrant colesevelam glucose sensor (8), capable of improving improves glycemic control. glucose tolerance by promoting glucose RESEARCH DESIGN AND METHODSdWe performed a frequently sampled intrave- utilization and triglyceride synthesis and nous glucose tolerance test (FSIGT) with minimal model analysis and a meal tolerance test (MTT) inhibiting gluconeogenesis (9,10). We in 20 subjects with impaired fasting glucose (11 men, 9 women; mean age 60.7 6 1.9 years, BMI hypothesized that independent of the 29.4 6 0.9 kg/m2) in a single-blind study after 2 weeks of placebo treatment and 8 weeks of precise mechanism of the effects of bile colesevelam 3.75 g daily. From these tests, insulin sensitivity, b-cell function, and glucose acid sequestrants on glucose, if the FXR- tolerance were determined, along with gastrointestinal peptide levels during the MTT. LXR hypothesis is correct, treatment of humans with colesevelam would result RESULTSdFasting plasma glucose and HbA1c decreased with colesevelam (from 5.9 6 0.1 to 5.7 6 0.1 mmol/L, P , 0.05, and from 5.86 6 0.06 to 5.76 6 0.06%, P = 0.01, respectively), but in improvement in insulin sensitivity. -

Retroviral-Mediated Gene Transfer and Expression of Human
Proc. Nati. Acad. Sci. USA Vol. 85, pp. 8146-8150, November 1988 Genetics Retroviral-mediated gene transfer and expression of human phenylalanine hydroxylase in primary mouse hepatocytes HONG PENG*, DONNA ARMENTANO*t, LESLIE MACKENZIE-GRAHAM*t, RONG-FONG SHEN*t, GRETCHEN DARLINGTONt, FRED D. LEDLEY*t, AND SAVIo L. C. WOO*t Departments of *Cell Biology and tPathology and Institute for Molecular Genetics, tHoward Hughes Medical Institute, Baylor College of Medicine, Houston, TX 77030 Communicated by Earl W. Davie, July 27, 1988 (received for review April 20, 1988) ABSTRACT Genetic therapy for phenylketonuria (severe infection of primary mouse hepatocytes with recombinant phenylalanine hydroxylase deficiency) may require introduc- retroviruses containing a neomycin-resistance marker gene. tion of a normal phenylalanine hydroxylase gene into hepatic Primary hepatocytes infected with the recombinant retrovi- cells of patients. We report development of a recombinant ruses were transformed to neomycin resistance and continue retrovirus based on the N2 vector for gene transfer and ex- to express liver-specific function (9). pression of human phenylalanine hydroxylase cDNA in primary We now report construction of a recombinant retrovirus mouse hepatocytes. This construct contains an internal pro- containing the human PAH cDNA under the control of moter ofthe human a1-antitrypsin gene driving transcription of transcriptional regulatory elements of the human a1- the phenylalanine hydroxylase cDNA. Primary mouse hepa- antitrypsin (a1AT) gene. These promoter elements have been tocytes were isolated from newborn mice, infected with the shown to direct high-level tissue-specific expression of a recombinant virus, and selected for expression ofthe neomycin- chloramphenicol acetyltransferase marker gene in cultured resistance gene. -

Engineering a Seven Enzyme Biotransformation Using
bioRxiv preprint doi: https://doi.org/10.1101/603795; this version posted April 9, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Engineering a seven enzyme biotransformation using mathematical modelling and characterized enzyme parts William Finnigan1,†, Rhys Cutlan2, Radka Snajdrova3,‡, Joseph P. Adams3, Jennifer A. Littlechild1 and Nicholas J. Harmer1,2* 1Department of Biosciences, Henry Wellcome Building for Biocatalysis, Stocker Road, Exeter EX4 4QD, U.K. 2Living Systems Institute, Stocker Road, Exeter EX4 4QD, U.K. 3GlaxoSmithKline R&D Ltd., Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, U.K. *Corresponding author: Nicholas J. Harmer, Living Systems Institute, Stocker Road, Exeter EX4 4QD, U.K., tel: +44 1392 725179, e-mail: [email protected] Keywords: Enzyme cascade, Kinetics, Cofactor recycling, Pathway optimization, Isolated enzymes, Reaction engineering, in vitro biocatalysis. Abstract Multi-step enzyme reactions offer considerable cost and productivity benefits. Process models offer a route to understanding the complexity of these reactions, and allow for their optimization. Despite the increasing prevalence of multi-step biotransformations, there are few examples of process models for enzyme reactions. From a toolbox of characterized enzyme parts, we demonstrate the construction of a process model for a seven enzyme, three step biotransformation using isolated enzymes. Enzymes for cofactor regeneration were employed to make this in vitro reaction economical. Good modelling practice was critical in evaluating the impact of approximations and experimental error. -

The Roles of OVATE and Other Elongation Genes in Regulating Proximal-Distal Patterning of Tomato Fruit
The roles of OVATE and other elongation genes in regulating proximal-distal patterning of tomato fruit Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Shan Wu, M.S. Graduate Program in Horticulture and Crop Science The Ohio State University 2015 Dissertation Committee: Dr. Esther van der Knaap, Advisor Dr. Jyan-chyun Jang Dr. Michelle Jones Dr. Tea Meulia Dr. Eric Stockinger Copyright by Shan Wu 2015 Abstract Domestication of tomato (Solanum lycopersicum) has resulted in a variety of fruit shapes from flat to very elongated. The elongated shape is a common feature that distinguishes many cultivated tomatoes from the undomesticated types, and it is mainly controlled by three loci, ovate, sun and fs8.1. We performed detailed morphological analyses of the reproductive and vegetative organs, which demonstrated that ovate, sun and fs8.1 regulate unique aspects of ovary and fruit elongation and in different temporal manners. The synergistic interaction between sun and ovate or fs8.1 suggested that the pathways involving SUN, OVATE and the gene(s) underlying fs8.1 may converge at a common node. We also conducted a transcriptome comparison analysis between the triple mutant sun/ovate/fs8.1 and wild type by RNA-seq using reproductive meristems and young flower buds. This study revealed changes in the transcription profile possibly caused by the combined effects of the three mutations. To gain insights into the role of OVATE in regulating fruit shape, we searched for its genetic and protein interactors. A synergistic interaction was found between ovate and suppressor of ovate 1 (sov1) in controlling ovary and fruit elongation.