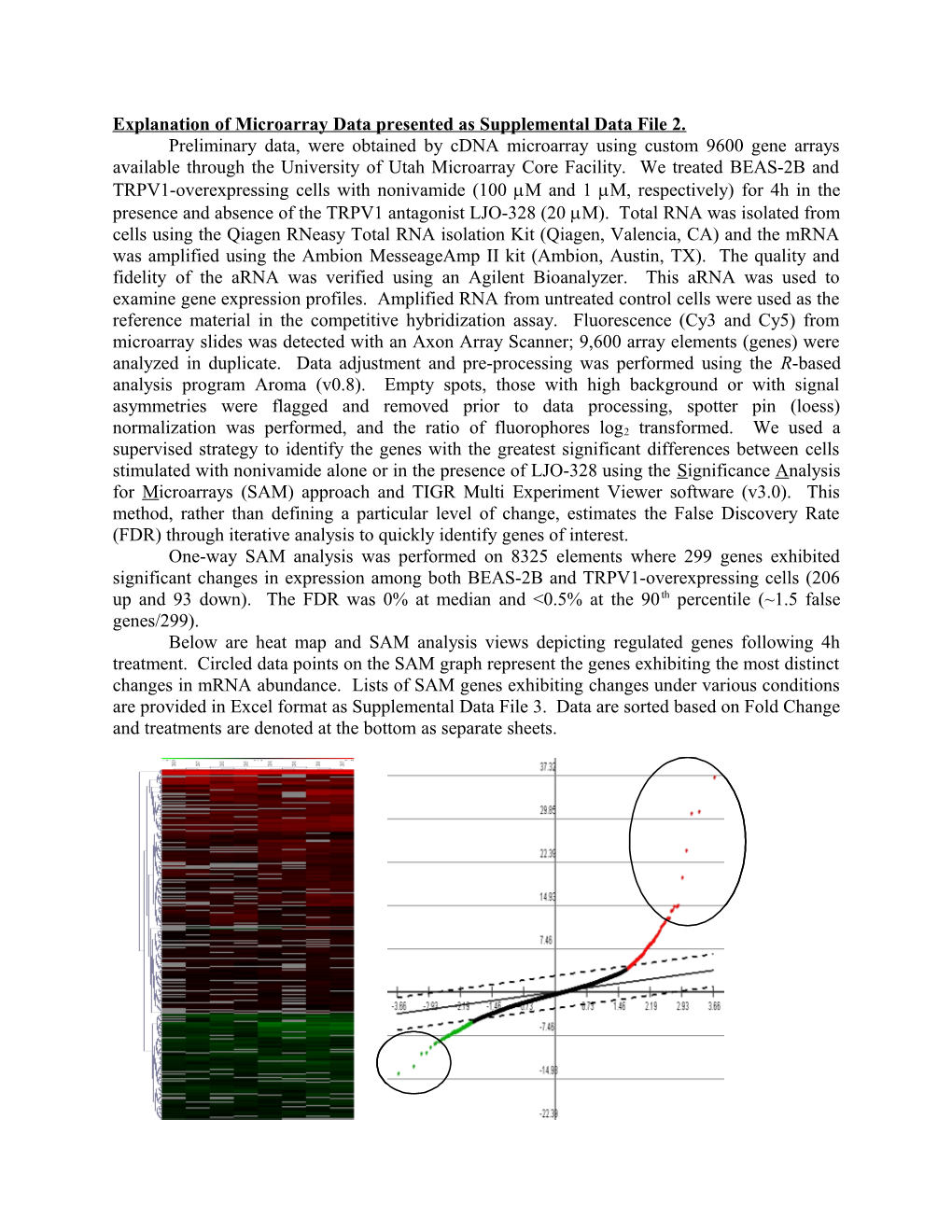
Explanation of Microarray Data presented as Supplemental Data File 2. Preliminary data, were obtained by cDNA microarray using custom 9600 gene arrays available through the University of Utah Microarray Core Facility. We treated BEAS-2B and TRPV1-overexpressing cells with nonivamide (100 M and 1 M, respectively) for 4h in the presence and absence of the TRPV1 antagonist LJO-328 (20 M). Total RNA was isolated from cells using the Qiagen RNeasy Total RNA isolation Kit (Qiagen, Valencia, CA) and the mRNA was amplified using the Ambion MesseageAmp II kit (Ambion, Austin, TX). The quality and fidelity of the aRNA was verified using an Agilent Bioanalyzer. This aRNA was used to examine gene expression profiles. Amplified RNA from untreated control cells were used as the reference material in the competitive hybridization assay. Fluorescence (Cy3 and Cy5) from microarray slides was detected with an Axon Array Scanner; 9,600 array elements (genes) were analyzed in duplicate. Data adjustment and pre-processing was performed using the R-based analysis program Aroma (v0.8). Empty spots, those with high background or with signal asymmetries were flagged and removed prior to data processing, spotter pin (loess) normalization was performed, and the ratio of fluorophores log2 transformed. We used a supervised strategy to identify the genes with the greatest significant differences between cells stimulated with nonivamide alone or in the presence of LJO-328 using the Significance Analysis for Microarrays (SAM) approach and TIGR Multi Experiment Viewer software (v3.0). This method, rather than defining a particular level of change, estimates the False Discovery Rate (FDR) through iterative analysis to quickly identify genes of interest. One-way SAM analysis was performed on 8325 elements where 299 genes exhibited significant changes in expression among both BEAS-2B and TRPV1-overexpressing cells (206 up and 93 down). The FDR was 0% at median and <0.5% at the 90 th percentile (~1.5 false genes/299). Below are heat map and SAM analysis views depicting regulated genes following 4h treatment. Circled data points on the SAM graph represent the genes exhibiting the most distinct changes in mRNA abundance. Lists of SAM genes exhibiting changes under various conditions are provided in Excel format as Supplemental Data File 3. Data are sorted based on Fold Change and treatments are denoted at the bottom as separate sheets.
Explanation of Microarray Data Presented As Supplemental Data File 2
Total Page:16
File Type:pdf, Size:1020Kb
Recommended publications