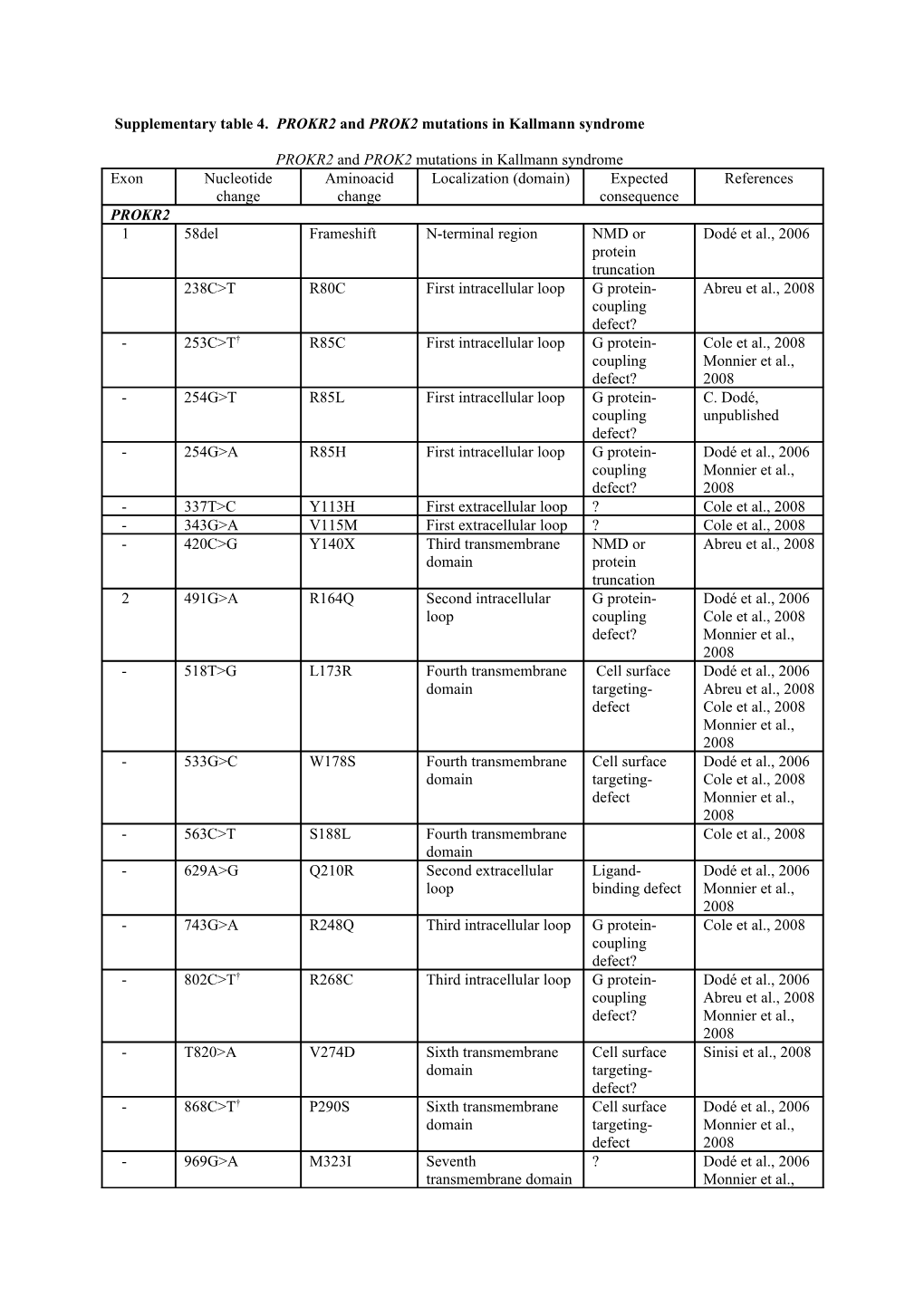
Supplementary table 4. PROKR2 and PROK2 mutations in Kallmann syndrome
PROKR2 and PROK2 mutations in Kallmann syndrome Exon Nucleotide Aminoacid Localization (domain) Expected References change change consequence PROKR2 1 58del Frameshift N-terminal region NMD or Dodé et al., 2006 protein truncation 238C>T R80C First intracellular loop G protein- Abreu et al., 2008 coupling defect? - 253C>T† R85C First intracellular loop G protein- Cole et al., 2008 coupling Monnier et al., defect? 2008 - 254G>T R85L First intracellular loop G protein- C. Dodé, coupling unpublished defect? - 254G>A R85H First intracellular loop G protein- Dodé et al., 2006 coupling Monnier et al., defect? 2008 - 337T>C Y113H First extracellular loop ? Cole et al., 2008 - 343G>A V115M First extracellular loop ? Cole et al., 2008 - 420C>G Y140X Third transmembrane NMD or Abreu et al., 2008 domain protein truncation 2 491G>A R164Q Second intracellular G protein- Dodé et al., 2006 loop coupling Cole et al., 2008 defect? Monnier et al., 2008 - 518T>G L173R Fourth transmembrane Cell surface Dodé et al., 2006 domain targeting- Abreu et al., 2008 defect Cole et al., 2008 Monnier et al., 2008 - 533G>C W178S Fourth transmembrane Cell surface Dodé et al., 2006 domain targeting- Cole et al., 2008 defect Monnier et al., 2008 - 563C>T S188L Fourth transmembrane Cole et al., 2008 domain - 629A>G Q210R Second extracellular Ligand- Dodé et al., 2006 loop binding defect Monnier et al., 2008 - 743G>A R248Q Third intracellular loop G protein- Cole et al., 2008 coupling defect? - 802C>T† R268C Third intracellular loop G protein- Dodé et al., 2006 coupling Abreu et al., 2008 defect? Monnier et al., 2008 - T820>A V274D Sixth transmembrane Cell surface Sinisi et al., 2008 domain targeting- defect? - 868C>T† P290S Sixth transmembrane Cell surface Dodé et al., 2006 domain targeting- Monnier et al., defect 2008 - 969G>A M323I Seventh ? Dodé et al., 2006 transmembrane domain Monnier et al., 2008 - 991G>A† V331M Seventh G protein- Dodé et al., 2006 transmembrane domain coupling Cole et al., 2008 defect? Monnier et al., 2008 - 1069C>T R357W C-terminal region G protein- Cole et al., 2008 coupling defect? PROK2 1 -4C>A Translation initiation Reduced Dodé et al., 2006 site protein synthesis - 70G>C A24P Signal peptide Targeting Cole et al., 2008 defect - 94G>C G32R AVITGA motif Loss of protein Dodé et al., 2006 activity 2 101G>A C34Y Cysteine-rich region Disulfide bond Cole et al., 2008 loss - 150C>G I50M Cysteine-rich region ? Cole et al., 2008 - 161G>A S54N Cysteine-rich region ? C. Dodé, unpublished - 163del Frameshift Cysteine-rich region NMD or Pitteloud et al., protein 2007 truncation Leroy et al., 2008 - 217C>T R73C Cysteine-rich region Disulfide bond Dodé et al., 2006 disruption Leroy et al., 2008 Cole et al., 2008 4 297_298insT Frameshift Cysteine-rich region NMD or Dodé et al., 2006 protein Abreu et al, 2008 truncation - 310C>T H104Y Cysteine-rich region ? C. Dodé, unpublished
Mutations reported in PROKR2 and PROK2 are mainly missense mutations. In most patients, the mutations have been found in heterozygous state. The p.R85H and p.L173R PROKR2 mutations, as well as p.R73C, c.163del, and c.297_298insT PROK2 mutations, however, have been found both in heterozygous and homozygous (or compound heterozygous) states, which suggests that patients heterozygous for PROKR2 or PROK2 mutations carry additional mutations, presumably in other as yet unidentified Kallmann syndrome genes in most cases. Notably, one such patient has the p.R85L mutation in PROKR2 together with a p.A604T mutation in FGFR1 (C. Dodé, unpublished), another patient has the p.V115M mutation in PROKR2 together with the p.A24P mutation in PROK2 (Cole et al., 2008), and a third patient has the p.L173R mutation in PROKR2 together with a p.S396L mutation in KAL1 (Dodé et al., 2006). † This mutation has also been found in one out of 250 individuals from the general population. Abbreviation: NMD, nonsense-mediated mRNA decay. References
Abreu A, Trarbach E, de Castro M, et al. (2008) Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J Clin Endocrinol Metab 10: 4113–4118.
Cole LW, Sidis Y, Zhang C, et al. (2008) Mutations in prokineticin 2 (PROK2) and PROK2 receptor (PROKR2) in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab 93: 3551–3559.
Dodé C, Teixeira L, Levilliers J, et al. (2006) Kallmann syndrome: mutations in the genes encoding prokineticin- 2 and prokineticin receptor-2. PLoS Genet 2: 1648–1652.
Monnier C, Dodé C, Fabre L, et al. (2008) PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling-activity. Hum Mol Genet (in press).
Pitteloud N, Zhang C, Pignatelli D, et al. (2007) Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A 104: 17447–17452.
Leroy C, Fouveaut C, Leclercq S, et al. (2008) Biallelic mutations in the prokineticin-2 gene in two sporadic cases of Kallmann syndrome. Eur J Hum Genet 16: 865–868.
Sinisi AA, Asci R, Bellastella G, et al. (2008) Homozygous mutation in the prokineticin-receptor 2 gene (Val274Asp) presenting as reversible Kallmann syndrome and persistent oligozoospermia: case report. Hum Reprod 23: 2380–2384.