Maf1 Suppression of ATF5-Dependent Mitochondrial Unfolded Protein Response Contributes to Rapamycin-Induced Radio-Sensitivity in Lung Cancer Cell Line A549
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 the Neuroprotective Transcription Factor ATF5 Is Decreased And
The neuroprotective transcription factor ATF5 is decreased and sequestered into polyglutamine inclusions in Huntington’s disease Ivó H. Hernández1,2,3, Jesús Torres-Peraza1,2,5, María Santos-Galindo1,2, Eloísa Ramos-Morón4, María R. Fernández-Fernández1,2, María J. Pérez-Álvarez1,2,3, Antonio Miranda-Vizuete4 and José J. Lucas1,2* 1 Centro de Biología Molecular Severo Ochoa (CBM”SO”) CSIC/UAM, Madrid, Spain. 2 Instituto de Salud Carlos III, Networking Research Center on Neurodegenerative Diseases (CIBERNED), Spain. 3 Departamento de Biología, Facultad de Ciencias, Universidad Autónoma de Madrid, Madrid, Spain. 4 Instituto de Biomedicina de Sevilla, Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, 41013 Sevilla, Spain 5 Present Address: Gerència d’Atenció Primària del Servei de Salut de les Illes Balears (IB-SALUT), Palma, Spain *Corresponding author: José J. Lucas [email protected] http://www.cbm.uam.es/lineas/lucasgroup.htm provided by Digital.CSIC View metadata, citation and similar papers at core.ac.uk CORE brought to you by 1 Abstract Activating transcription factor-5 (ATF5) is a stress-response transcription factor induced upon different cell stressors like fasting, amino-acid limitation, cadmium or arsenite. ATF5 is also induced, and promotes transcription of anti-apoptotic target genes like MCL1, during the unfolded protein response (UPR) triggered by endoplasmic reticulum stress. In the brain, high ATF5 levels are found in gliomas and also in neural progenitor cells, which need to decrease their ATF5 levels for differentiation into mature neurons or glia. This initially led to believe that ATF5 is not expressed in adult neurons. More recently, we reported basal neuronal ATF5 expression in adult mouse brain and its neuroprotective induction during UPR in a mouse model of status epilepticus. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

ATF3 Negatively Regulates Cellular Antiviral Signaling and Autophagy In
www.nature.com/scientificreports OPEN ATF3 negatively regulates cellular antiviral signaling and autophagy in the absence of type I interferons Received: 21 April 2017 Vikas Sood1, Kiran Bala Sharma1, Vishal Gupta1, Dhurjhoti Saha2, Parashar Dhapola2,4, Accepted: 21 July 2017 Manish Sharma1, Utsav Sen1, Shigetaka Kitajima3, Shantanu Chowdhury2,4, Manjula Kalia 1 Published: xx xx xxxx & Sudhanshu Vrati1,5 Stringent regulation of antiviral signaling and cellular autophagy is critical for the host response to virus infection. However, little is known how these cellular processes are regulated in the absence of type I interferon signaling. Here, we show that ATF3 is induced following Japanese encephalitis virus (JEV) infection, and regulates cellular antiviral and autophagy pathways in the absence of type I interferons in mouse neuronal cells. We have identifed new targets of ATF3 and show that it binds to the promoter regions of Stat1, Irf9, Isg15 and Atg5 thereby inhibiting cellular antiviral signaling and autophagy. Consistent with these observations, ATF3-depleted cells showed enhanced antiviral responses and induction of robust autophagy. Furthermore, we show that JEV replication was signifcantly reduced in ATF3-depleted cells. Our fndings identify ATF3 as a negative regulator of antiviral signaling and cellular autophagy in mammalian cells, and demonstrate its important role in JEV life cycle. Viruses are arduous pathogens that pose a unique challenge to our immune system as they are composed of the host-derived molecules. However, viral nucleic acids possess unique features distinguishing them from the host which have possibly led to the evolution of Pattern Recognition Receptors (PRRs) for their detection. Among the PRRs, RIG-I-like receptors (RLRs) are ubiquitous cytosolic detectors which play an integral role in antiviral responses1. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Cell Type-Dependent Function of ATF5: Where We Go from Here Kathleen L
lopmen ve ta e l B D Lengel and Liu, Cell Dev Biol 2012, 1:5 io & l l o DOI: 10.4172/2168-9296.1000e114 l g e y C Cell & Developmental Biology ISSN: 2168-9296 Editorial Open Access Cell Type-Dependent Function of ATF5: Where We Go from Here Kathleen L. Lengel and David X. Liu* Penn State College of Medicine, 500 University Drive, Hershey, PA 17033, USA Uncontrollable cell proliferation and suppressed apoptosis are Concluding Remarks hallmarks of tumorigenic transformation [1-3]. Deregulation of genes controlling cell proliferation and survival plays an important role in the Current research indicates that ATF5 plays important roles in process. For instance, apoptotic genes are frequently down-regulated diverse biological processes including cell proliferation, cell survival, while anti-apoptotic genes are highly expressed in a number of cancer cellular stress response, cell fate determination, and is involved in the cell types; artificial down regulation of anti-apoptotic genes or up development of several human diseases such as cancer and neurological regulation of apoptotic genes are often sufficient to eradicate those disorders. Our understanding of the mechanism by which ATF5 cancer cells. The activating transcription factor 5 (ATF5 or ATFx) is functions is very limited and many questions remain unanswered. a member of the ATF/CREB transcription factor family. Although There are a number of urgent questions need to be addressed. For ATF5 is known to regulate cell cycle progression [4-7], cell survival instance, what are the causes -

Combinatorial Bzip Dimers Display Complex DNA-Binding Specificity Landscapes
Combinatorial bZIP dimers display complex DNA-binding specificity landscapes The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Rodriguez-Martinez, Jose A et al. “Combinatorial bZIP Dimers Display Complex DNA-Binding Specificity Landscapes.” eLife 6 (2017): n. pag. As Published http://dx.doi.org/10.7554/eLife.19272 Publisher eLife Sciences Publications, Ltd. Version Final published version Citable link http://hdl.handle.net/1721.1/110147 Terms of Use Creative Commons Attribution 4.0 International License Detailed Terms http://creativecommons.org/licenses/by-nc/4.0/ RESEARCH ARTICLE Combinatorial bZIP dimers display complex DNA-binding specificity landscapes Jose´ A Rodrı´guez-Martı´nez1†, Aaron W Reinke2†, Devesh Bhimsaria1,3†, Amy E Keating2,4, Aseem Z Ansari1,5* 1Department of Biochemistry, University of Wisconsin-Madison, Madison, United States; 2Department of Biology, Massachusetts Institute of Technology, Cambridge, United States; 3Department of Electrical and Computer Engineering, University of Wisconsin-Madison, Madison, Unites States; 4Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, United States; 5The Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, United States Abstract How transcription factor dimerization impacts DNA-binding specificity is poorly understood. Guided by protein dimerization properties, we examined DNA binding specificities of 270 human bZIP pairs. DNA interactomes of 80 heterodimers and 22 homodimers revealed that 72% of heterodimer motifs correspond to conjoined half-sites preferred by partnering monomers. Remarkably, the remaining motifs are composed of variably-spaced half-sites (12%) or ‘emergent’ sites (16%) that cannot be readily inferred from half-site preferences of partnering monomers. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Supplementary Table 5. Clover Results Indicate the Number Of
Supplementary Table 5. Clover results indicate the number of chromosomes with transcription factor binding motifs statistically over‐ or under‐represented in HTE DHS within intergenic sequence (more than 2kb outside of any gene). Analysis was divided into three groups (all DHS, HTE‐selective DHS, and ubiquitous DHS). Motifs with more than one entry in the databases utilized were edited to retain only the first occurrence of the motif. All DHS x Intergenic TEselective DHS x Intergenic Ubiquitous DHS x Intergenic ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 MA0002.2 RUNX1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0003.1 TFAP2A 23 0 MA0089.1 NFE2L1::MafG 23 0 MA0068.1 Pax4 23 0 MA0039.2 Klf4 23 0 MA0098.1 ETS1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0099.2 AP1 23 0 MA0098.1 ETS1 23 0 MA0056.1 MZF1_1‐4 23 0 MA0136.1 ELF5 23 0 MA0139.1 CTCF 23 0 MA0079.2 SP1 23 0 MA0145.1 Tcfcp2l1 23 0 V$ALX3_01 ALX‐3 23 0 MA0080.2 SPI1 23 0 MA0150.1 NFE2L2 23 0 V$ALX4_02 Alx‐4 23 0 myocyte enhancer MA0081.1 SPIB 23 0 MA0156.1 FEV 23 0 V$AMEF2_Q6 factor 23 0 MA0089.1 NFE2L1::MafG 23 0 V$AP1FJ_Q2 activator protein 1 23 0 V$AP1_01 AP‐1 23 0 MA0090.1 TEAD1 23 0 V$AP4_Q5 activator protein 4 23 0 V$AP2_Q6_01 AP‐2 23 0 MA0098.1 ETS1 23 0 V$AR_Q6 half‐site matrix 23 0 V$ARX_01 Arx 23 0 BTB and CNC homolog MA0099.2 AP1 23 0 V$BACH1_01 1 23 0 V$BARHL1_01 Barhl‐1 23 0 BTB and CNC homolog MA0136.1 ELF5 23 0 V$BACH2_01 2 23 0 V$BARHL2_01 Barhl2 23 0 MA0139.1 CTCF 23 0 V$CMAF_02 C‐MAF 23 0 V$BARX1_01 Barx1 23 0 MA0144.1 Stat3 23 0 -

Profiling Transcription Factor Sub-Networks in Type I Interferon Signaling and in Response To
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.25.428122; this version posted January 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Profiling transcription factor sub-networks in type I interferon signaling and in response to SARS-CoV-2 infection * Chilakamarti V. Ramana ORCID ID: /0000-0002-5153-8252 *Correspondence should be addressed: Chilakamarti V .Ramana, E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.25.428122; this version posted January 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Type I interferons (IFN α/β) play a central role in innate immunity to respiratory viruses, including coronaviruses. Genetic defects in type I interferon signaling were reported in a significant proportion of critically ill CoOVID-19 patients. Extensive studies on interferon-induced intracellular signal transduction pathways led to the elucidation of the Jak-Stat pathway. Furthermore, advances in gene expression profiling by microarrays have revealed that type I interferon rapidly induced multiple transcription factor mRNA levels. In this study, transcription factor profiling in the transcriptome was used to gain novel insights into the role of inducible transcription factors in response to type I interferon signaling in immune cells and in lung epithelial cells after SARS-CoV-2 infection. -

Transcription Factor ATF5 Is Required for Terminal Differentiation and Survival of Olfactory Sensory Neurons
Transcription factor ATF5 is required for terminal differentiation and survival of olfactory sensory neurons Shu-Zong Wanga,b, Jianhong Oub, Lihua J. Zhub,c, and Michael R. Greena,b,1 aHoward Hughes Medical Institute, bPrograms in Gene Function and Expression and Molecular Medicine, and cProgram in Bioinformatics and Integrative Biology, University of Massachusetts Medical School, Worcester, MA 01605 Edited by Solomon H. Snyder, The Johns Hopkins University School of Medicine, Baltimore, MD, and approved September 27, 2012 (received for review June 21, 2012) Activating transcription factor 5 (ATF5) is a member of the ATF/cAMP not been determined. Here we address this issue by deriving and −/− response element-binding family of transcription factors, which characterizing Atf5 mice. compose a large group of basic region leucine zipper proteins whose members mediate diverse transcriptional regulatory functions. Results −/− ATF5 has a well-established prosurvival activity and has been found Derivation of Atf5 Mice. To investigate the physiological role of −/− to be overexpressed in several human cancers, in particular glioblas- ATF5, we derived Atf5 mice. The gene-targeting strategy in- toma. However, the role(s) of ATF5 in development and normal volved replacing almost the entire coding region of Atf5 with a physiology are unknown. Here we address this issue by deriving LacZ-Neo cassette, such that the LacZ gene is under the control and characterizing homozygous Atf5 knockout mice. We find that of the endogenous Atf5 promoter (Fig. S1A). The genomic struc- +/+ +/− −/− Atf5−/− pups die neonatally, which, as explained below, is consis- ture of the Atf5 locus in Atf5 , Atf5 ,andAtf5 mice was fi tent with an olfactory defect resulting in a competitive suckling con rmed by Southern blot (Fig. -
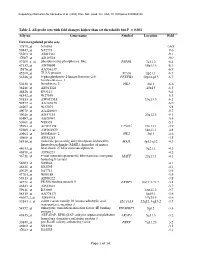
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Proteotoxic Stress and Cell Death in Cancer Cells
cancers Review Proteotoxic Stress and Cell Death in Cancer Cells Claudio Brancolini * and Luca Iuliano Department of Medicine, Università degli Studi di Udine, p.le Kolbe 4—33100 Udine, Italy; [email protected] * Correspondence: [email protected] Received: 14 July 2020; Accepted: 20 August 2020; Published: 23 August 2020 Abstract: To maintain proteostasis, cells must integrate information and activities that supervise protein synthesis, protein folding, conformational stability, and also protein degradation. Extrinsic and intrinsic conditions can both impact normal proteostasis, causing the appearance of proteotoxic stress. Initially, proteotoxic stress elicits adaptive responses aimed at restoring proteostasis, allowing cells to survive the stress condition. However, if the proteostasis restoration fails, a permanent and sustained proteotoxic stress can be deleterious, and cell death ensues. Many cancer cells convive with high levels of proteotoxic stress, and this condition could be exploited from a therapeutic perspective. Understanding the cell death pathways engaged by proteotoxic stress is instrumental to better hijack the proliferative fate of cancer cells. Keywords: UPR; NOXA; DR5; BCL2; apoptosis; necroptosis; ferroptosis; proteotoxic stress 1. Proteotoxic Stress: An Introduction Proteins are key macromolecules that play fundamental roles in almost every cellular process, from gene expression to cell/tissue protection [1]. The important and relentless actions of proteins oblige cells to supervise and guarantee their correct folding and assembling. Protein homeostasis or proteostasis is the fundamental cellular effort aimed at reaching this goal. Proteostasis is governed through a complex network of regulative mechanisms and is an essential task for cell survival [2]. The vast majority of proteins need to assume a peculiar thermodynamically stable three-dimensional structure that depends on their amino acid sequence [3].