I. Infants with Poor Vision
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Differentiate Red Eye Disorders
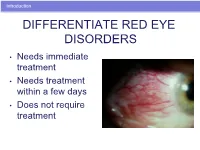
Introduction DIFFERENTIATE RED EYE DISORDERS • Needs immediate treatment • Needs treatment within a few days • Does not require treatment Introduction SUBJECTIVE EYE COMPLAINTS • Decreased vision • Pain • Redness Characterize the complaint through history and exam. Introduction TYPES OF RED EYE DISORDERS • Mechanical trauma • Chemical trauma • Inflammation/infection Introduction ETIOLOGIES OF RED EYE 1. Chemical injury 2. Angle-closure glaucoma 3. Ocular foreign body 4. Corneal abrasion 5. Uveitis 6. Conjunctivitis 7. Ocular surface disease 8. Subconjunctival hemorrhage Evaluation RED EYE: POSSIBLE CAUSES • Trauma • Chemicals • Infection • Allergy • Systemic conditions Evaluation RED EYE: CAUSE AND EFFECT Symptom Cause Itching Allergy Burning Lid disorders, dry eye Foreign body sensation Foreign body, corneal abrasion Localized lid tenderness Hordeolum, chalazion Evaluation RED EYE: CAUSE AND EFFECT (Continued) Symptom Cause Deep, intense pain Corneal abrasions, scleritis, iritis, acute glaucoma, sinusitis, etc. Photophobia Corneal abrasions, iritis, acute glaucoma Halo vision Corneal edema (acute glaucoma, uveitis) Evaluation Equipment needed to evaluate red eye Evaluation Refer red eye with vision loss to ophthalmologist for evaluation Evaluation RED EYE DISORDERS: AN ANATOMIC APPROACH • Face • Adnexa – Orbital area – Lids – Ocular movements • Globe – Conjunctiva, sclera – Anterior chamber (using slit lamp if possible) – Intraocular pressure Disorders of the Ocular Adnexa Disorders of the Ocular Adnexa Hordeolum Disorders of the Ocular -

Peripapillary Retinal Vascular Involvement in Early Post-COVID-19 Patients
Journal of Clinical Medicine Article Peripapillary Retinal Vascular Involvement in Early Post-COVID-19 Patients 1,2, 1,2, 1,2, Alfonso Savastano y , Emanuele Crincoli y , Maria Cristina Savastano * , Saad Younis 3, Gloria Gambini 1,2, Umberto De Vico 1,2 , Grazia Maria Cozzupoli 1,2 , Carola Culiersi 1,2 , Stanislao Rizzo 1,2,4 and Gemelli Against COVID-19 Post-Acute Care Study Group 2 1 Ophthalmology Unit, Fondazione Policlinico Universitario A. Gemelli IRCCS, 00196 Rome, Italy; [email protected] (A.S.); [email protected] (E.C.); [email protected] (G.G.); [email protected] (U.D.V.); [email protected] (G.M.C.); [email protected] (C.C.); [email protected] (S.R.) 2 Department of Ophthalmology, Catholic University of “Sacro Cuore”, 00168 Rome, Italy 3 Department of Ophthalmology, Western Eye Hospital, Imperial College Healthcare NHS Trust, London NW1 5QH, UK; [email protected] 4 Neuroscience Institute, Consiglio Nazionale delle Ricerche, Istituto di Neuroscienze, 56124 Pisa, Italy * Correspondence: [email protected]; Tel.: +39-063-015-4928 These authors contributed equally to this work. y Received: 5 August 2020; Accepted: 3 September 2020; Published: 8 September 2020 Abstract: The ability of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-20s) to cause multi-organ ischemia and coronavirus-induced posterior segment eye diseases in mammals gave concern about potential sight-threatening ischemia in post coronavirus disease 2019 patients. The radial peripapillary capillary plexus (RPCP) is a sensitive target due to the important role in the vascular supply of the peripapillary retinal nerve fiber layer (RNFL). -

Olivia Steinberg ICO Primary Care/Ocular Disease Resident American Academy of Optometry Residents Day Submission
Olivia Steinberg ICO Primary Care/Ocular Disease Resident American Academy of Optometry Residents Day Submission The use of oral doxycycline and vitamin C in the management of acute corneal hydrops: a case comparison Abstract- We compare two patients presenting to clinic with an uncommon complication of keratoconus, acute corneal hydrops. Management of the patients differs. One heals quickly, while the other has a delayed course to resolution. I. Case A a. Demographics: 40 yo AAM b. Case History i. CC: red eye, tearing, decreased VA x 1 day OS ii. POHx: (+) keratoconus OU iii. PMHx: depression, anxiety, asthma iv. Meds: Albuterol, Ziprasidone v. Scleral CL wearer for approximately 6 months OU vi. Denies any pain OS, denies previous occurrence OU, no complaints OD c. Pertinent Findings i. VA cc (CL’s)- 20/25 OD, 20/200 PH 20/60+2 OS ii. Slit Lamp 1. Inferior corneal thinning and Fleisher ring OD, central scarring OD, 2+ diffuse microcystic edema OS, Descemet’s break OS (photos and anterior segment OCT) 2. 2+ diffuse injection OS 3. D&Q A/C OU iii. Intraocular Pressures: deferred OD due to CL, 9mmHg OS (tonopen) iv. Fundus Exam- unremarkable OU II. Case B a. Demographics: 39 yo AAM b. Case History i. CC: painful, red eye, tearing, decreased VA x 1 day OS ii. POHx: unremarkable iii. PMHx: hypertension iv. Meds: unknown HTN medication v. Wears Soflens toric CL’s OU; reports previous doctor had difficulty achieving proper fit OU; denies diagnosis of keratoconus OU vi. Denies any injury OS, denies previous occurrence OU, no complaints OD c. -

Smoking and Eye Disease
Smoking and Eye Disease Here are some eye problems that are made worse Smoking and eye disease by smoking: Smoking tobacco (cigarettes, cigars or pipes) can cause lung disease, heart disease, cancer, and Dry eye. This is when your eyes do not have many other serious health problems. But did you enough—or the right kind of—tears. Smoking know that smoking can also harm your eyes? with dry eye will make your eyes more likely to feel scratchy, sting, burn or be red. Eye Words to Know Cataracts. If you smoke you are at increased risk for getting cataracts. A cataract is clouding of Retina: Layer of nerve cells lining the back your eye’s naturally clear lens. It causes blurry wall inside the eye. This layer senses light and vision and makes colors look dull, faded or sends signals to the brain so you can see. yellowish. Cataracts are removed in surgery. Macula: Small but important area in the center of the retina. You need the macula to Age-related macular degeneration (AMD). This clearly see details of objects in front of you. disease happens when a part of the retina called the macula is damaged. You lose your central Lens: Clear part of the eye behind the colored vision and cannot see fine details. But your iris. It helps to focus light on the retina (back peripheral (side) vision stays normal. Sometimes of the eye) so you can see. medicine or surgery can help certain people with Optic nerve: A nerve at the back of your AMD from getting worse. -

Bass – Glaucomatous-Type Field Loss Not Due to Glaucoma
Glaucoma on the Brain! Glaucomatous-Type Yes, we see lots of glaucoma Field Loss Not Due to Not every field that looks like glaucoma is due to glaucoma! Glaucoma If you misdiagnose glaucoma, you could miss other sight-threatening and life-threatening Sherry J. Bass, OD, FAAO disorders SUNY College of Optometry New York, NY Types of Glaucomatous Visual Field Defects Paracentral Defects Nasal Step Defects Arcuate and Bjerrum Defects Altitudinal Defects Peripheral Field Constriction to Tunnel Fields 1 Visual Field Defects in Very Early Glaucoma Paracentral loss Early superior/inferior temporal RNFL and rim loss: short axons Arcuate defects above or below the papillomacular bundle Arcuate field loss in the nasal field close to fixation Superotemporal notch Visual Field Defects in Early Glaucoma Nasal step More widespread RNFL loss and rim loss in the inferior or superior temporal rim tissue : longer axons Loss stops abruptly at the horizontal raphae “Step” pattern 2 Visual Field Defects in Moderate Glaucoma Arcuate scotoma- Bjerrum scotoma Focal notches in the inferior and/or superior rim tissue that reach the edge of the disc Denser field defects Follow an arcuate pattern connected to the blind spot 3 Visual Field Defects in Advanced Glaucoma End-Stage Glaucoma Dense Altitudinal Loss Progressive loss of superior or inferior rim tissue Non-Glaucomatous Etiology of End-Stage Glaucoma Paracentral Field Loss Peripheral constriction Hereditary macular Loss of temporal rim tissue diseases Temporal “islands” Stargardt’s macular due -

Optic Nerve Hypoplasia Plus: a New Way of Looking at Septo-Optic Dysplasia
Optic Nerve Hypoplasia Plus: A New Way of Looking at Septo-Optic Dysplasia Item Type text; Electronic Thesis Authors Mohan, Prithvi Mrinalini Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 29/09/2021 22:50:06 Item License http://rightsstatements.org/vocab/InC/1.0/ Link to Item http://hdl.handle.net/10150/625105 OPTIC NERVE HYPOPLASIA PLUS: A NEW WAY OF LOOKING AT SEPTO-OPTIC DYSPLASIA By PRITHVI MRINALINI MOHAN ____________________ A Thesis Submitted to The Honors College In Partial Fulfillment of the Bachelors degree With Honors in Physiology THE UNIVERSITY OF ARIZONA M A Y 2 0 1 7 Approved by: ____________________________ Dr. Vinodh Narayanan Center for Rare Childhood Disorders Abstract Septo-optic dysplasia (SOD) is a rare congenital disorder that affects 1/10,000 live births. At its core, SOD is a disorder resulting from improper embryological development of mid-line brain structures. To date, there is no comprehensive understanding of the etiology of SOD. Currently, SOD is diagnosed based on the presence of at least two of the following three factors: (i) optic nerve hypoplasia (ii) improper pituitary gland development and endocrine dysfunction and (iii) mid-line brain defects, including agenesis of the septum pellucidum and/or corpus callosum. A literature review of existing research on the disorder was conducted. The medical history and genetic data of 6 patients diagnosed with SOD were reviewed to find damaging variants. -

CAUSES, COMPLICATIONS &TREATMENT of A“RED EYE”
CAUSES, COMPLICATIONS & TREATMENT of a “RED EYE” 8 Most cases of “red eye” seen in general practice are likely to be conjunctivitis or a superficial corneal injury, however, red eye can also indicate a serious eye condition such as acute angle glaucoma, iritis, keratitis or scleritis. Features such as significant pain, photophobia, reduced visual acuity and a unilateral presentation are “red flags” that a sight-threatening condition may be present. In the absence of specialised eye examination equipment, such as a slit lamp, General Practitioners must rely on identifying these key features to know which patients require referral to an Ophthalmologist for further assessment. Is it conjunctivitis or is it something more Iritis is also known as anterior uveitis; posterior uveitis is serious? inflammation of the choroid (choroiditis). Complications include glaucoma, cataract and macular oedema. The most likely cause of a red eye in patients who present to 4. Scleritis is inflammation of the sclera. This is a very rare general practice is conjunctivitis. However, red eye can also be presentation, usually associated with autoimmune a feature of a more serious eye condition, in which a delay in disease, e.g. rheumatoid arthritis. treatment due to a missed diagnosis can result in permanent 5. Penetrating eye injury or embedded foreign body; red visual loss. In addition, the inappropriate use of antibacterial eye is not always a feature topical eye preparations contributes to antimicrobial 6. Acid or alkali burn to the eye resistance. The patient history will usually identify a penetrating eye injury Most general practice clinics will not have access to specialised or chemical burn to the eye, but further assessment may be equipment for eye examination, e.g. -

The Red Eye Differential Diagnosis Differential Diagnosis of “Red Eye”
The Red Eye Differential Diagnosis Differential Diagnosis of “red eye” Conjunctiva Pupil Cornea Anterior IOP chamber Subconjunctival Bright red Normal Normal Normal Normal Haemorrhage Conjunctivitis Injected Normal Normal Normal Normal vessels, fornices. Discharge Iritis Injected Small, Normal, Turgid, Normal around cornea fixed, KPs deep irregular Acute glaucoma Entire eye red Fixed, Hazy Shallow High dilated, oval Conjunctivitis Papillae Follicles Purulent discharge Redness Chemosis Subconjunctival Haemorrhage • Diffuse or localised area of blood under conjunctiva. Asymptomatic • Idiopathic, trauma, cough, sneezing, aspirin, HT • Resolves within 10-14 days Dry Eye Syndrome • Poor quality – Meibomian gland disease,Acne rosacea – Lid related – Vitamin A deficiency • Poor quantity –KCS • Sjogren Syndrome • Rheumatoid Arthritis – Lacrimal disease ie, Sarcoidosis – Paralytic ie, VII CN palsy Corneal Abrasion • Surface epithelium sloughed off. • Stains with fluorescein • Usually due to trauma • Pain, FB sensation, tearing, red eye Corneal Ulcer • Infection – Bacterial: Adnexal infection, lid malposition, dry eye, CL – Viral: HSV, HZO – Fungal: –Protozoan:Acanthamoeba in CL wearer • Mechanical or trauma • Chemical: Alkali injuries are worse than acid Episcleritis • Superficial • Idiopathic, collagen vascular disorder (RA) • Asymptomatic, mild pain • Self-limiting or topical treatment Scleritis •Deep • Idiopathic • Collagen vascular disease (RA,AS, SLE, Wegener, PAN) • Zoster • Sarcoidosis • Dull, deep pain wakes patient at night • Systemic -

TUBB3 M323V Syndrome Presents with Infantile Nystagmus
G C A T T A C G G C A T genes Case Report TUBB3 M323V Syndrome Presents with Infantile Nystagmus Soohwa Jin 1, Sung-Eun Park 2, Dongju Won 3, Seung-Tae Lee 3, Sueng-Han Han 2 and Jinu Han 4,* 1 Department of Opthalmology, Yonsei University College of Medicine, Seoul 03722, Korea; [email protected] 2 Department of Ophthalmology, Institute of Vision Research, Severance Hospital, Yonsei University College of Medicine, Seoul 03722, Korea; [email protected] (S.-E.P.); [email protected] (S.-H.H.) 3 Department of Laboratory Medicine, Severance Hospital, Yonsei University College of Medicine, Seoul 03722, Korea; [email protected] (D.W.); [email protected] (S.-T.L.) 4 Department of Ophthalmology, Institute of Vision Research, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul 06273, Korea * Correspondence: [email protected]; Tel.: +82-2-2019-3445 Abstract: Variants in the TUBB3 gene, one of the tubulin-encoding genes, are known to cause congenital fibrosis of the extraocular muscles type 3 and/or malformations of cortical development. Herein, we report a case of a 6-month-old infant with c.967A>G:p.(M323V) variant in the TUBB3 gene, who had only infantile nystagmus without other ophthalmological abnormalities. Subsequent brain magnetic resonance imaging (MRI) revealed cortical dysplasia. Neurological examinations did not reveal gross or fine motor delay, which are inconsistent with the clinical characteristics of patients with the M323V syndrome reported so far. A protein modeling showed that the M323V mutation in the TUBB3 gene interferes with αβ heterodimer formation with the TUBA1A gene. -

Canine Red Eye Elizabeth Barfield Laminack, DVM; Kathern Myrna, DVM, MS; and Phillip Anthony Moore, DVM, Diplomate ACVO
PEER REVIEWED Clinical Approach to the CANINE RED EYE Elizabeth Barfield Laminack, DVM; Kathern Myrna, DVM, MS; and Phillip Anthony Moore, DVM, Diplomate ACVO he acute red eye is a common clinical challenge for tion of the deep episcleral vessels, and is characterized general practitioners. Redness is the hallmark of by straight and immobile episcleral vessels, which run Tocular inflammation; it is a nonspecific sign related 90° to the limbus. Episcleral injection is an external to a number of underlying diseases and degree of redness sign of intraocular disease, such as anterior uveitis and may not reflect the severity of the ocular problem. glaucoma (Figures 3 and 4). Occasionally, episcleral Proper evaluation of the red eye depends on effective injection may occur in diseases of the sclera, such as and efficient diagnosis of the underlying ocular disease in episcleritis or scleritis.1 order to save the eye’s vision and the eye itself.1,2 • Corneal Neovascularization » Superficial: Long, branching corneal vessels; may be SOURCE OF REDNESS seen with superficial ulcerative (Figure 5) or nonul- The conjunctiva has small, fine, tortuous and movable vessels cerative keratitis (Figure 6) that help distinguish conjunctival inflammation from deeper » Focal deep: Straight, nonbranching corneal vessels; inflammation (see Ocular Redness algorithm, page 16). indicates a deep corneal keratitis • Conjunctival hyperemia presents with redness and » 360° deep: Corneal vessels in a 360° pattern around congestion of the conjunctival blood vessels, making the limbus; should arouse concern that glaucoma or them appear more prominent, and is associated with uveitis (Figure 4) is present1,2 extraocular disease, such as conjunctivitis (Figure 1). -

Multiple Ocular Developmental Defects in Four Closely Related Alpacas
Veterinary Ophthalmology (2018) 1–8 DOI:10.1111/vop.12540 CASE REPORT Multiple ocular developmental defects in four closely related alpacas Kelly E. Knickelbein,* David J. Maggs,§ Christopher M. Reilly,†,1 Kathryn L. Good*,2 and Juliet R. Gionfriddo‡,3 *The Veterinary Medical Teaching Hospital, University of California, Davis, CA 95616, USA; §Department of Surgical and Radiological Sciences, University of California, Davis, CA 95616 USA; †Department of Pathology Microbiology, and Immunology, University of California, Davis, CA 95616, USA; and ‡The College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO 80528, USA Address communications to: Abstract D. J. Maggs Objective To describe the clinical, gross pathologic, and histopathologic findings for a Tel.: (530) 752-3937 visually impaired 5.8-year-old female alpaca with multiple ocular abnormalities, as well Fax: (530) 752-6042 as the clinical findings for three closely related alpacas. e-mail: [email protected] Animals studied Four alpacas. Present addresses: 1Insight Procedures Ophthalmic examination was performed on a 16-month-old female alpaca Veterinary Specialty Pathology, following observation of visual impairment while hospitalized for an unrelated illness. Austin, TX 78752, USA Following acute systemic decline and death 4.5 years later, the alpaca’s brain, optic 2Department of Surgical and Radiological Sciences, nerves, and eyes were examined grossly and histologically. Ophthalmic examination of University of California, Davis, three closely related alpacas was subsequently performed. CA 95616, USA Results The 16-month-old female alpaca (Alpaca 1) had ophthalmoscopic findings sug- 3 Red Feather Lakes, CO 80545, gestive of a coloboma or hypoplasia of the retinal pigment epithelium (RPE) and chor- USA oid, and suspected optic nerve hypoplasia OU. -

Guidelines for Universal Eye Screening in Newborns Including RETINOPATHY of Prematurity
GUIDELINES FOR UNIVERSAL EYE SCREENING IN NEWBORNS INCLUDING RETINOPATHY OF PREMATURITY RASHTRIYA BAL SWASthYA KARYAKRAM Ministry of Health & Family Welfare Government of India June 2017 MESSAGE The Ministry of Health & Family Welfare, Government of India, under the National Health Mission launched the Rashtriya Bal Swasthya Karyakram (RBSK), an innovative and ambitious initiative, which envisages Child Health Screening and Early Intervention Services. The main focus of the RBSK program is to improve the quality of life of our children from the time of birth till 18 years through timely screening and early management of 4 ‘D’s namely Defects at birth, Development delays including disability, childhood Deficiencies and Diseases. To provide a healthy start to our newborns, RBSK screening begins at birth at delivery points through comprehensive screening of all newborns for various defects including eye and vision related problems. Some of these problems are present at birth like congenital cataract and some may present later like Retinopathy of prematurity which is found especially in preterm children and if missed, can lead to complete blindness. Early Newborn Eye examination is an integral part of RBSK comprehensive screening which would prevent childhood blindness and reduce visual and scholastic disabilities among children. Universal newborn eye screening at delivery points and at SNCUs provides a unique opportunity to identify and manage significant eye diseases in babies who would otherwise appear healthy to their parents. I wish that State and UTs would benefit from the ‘Guidelines for Universal Eye Screening in Newborns including Retinopathy of Prematurity’ and in supporting our future generation by providing them with disease free eyes and good quality vision to help them in their overall growth including scholastic achievement.