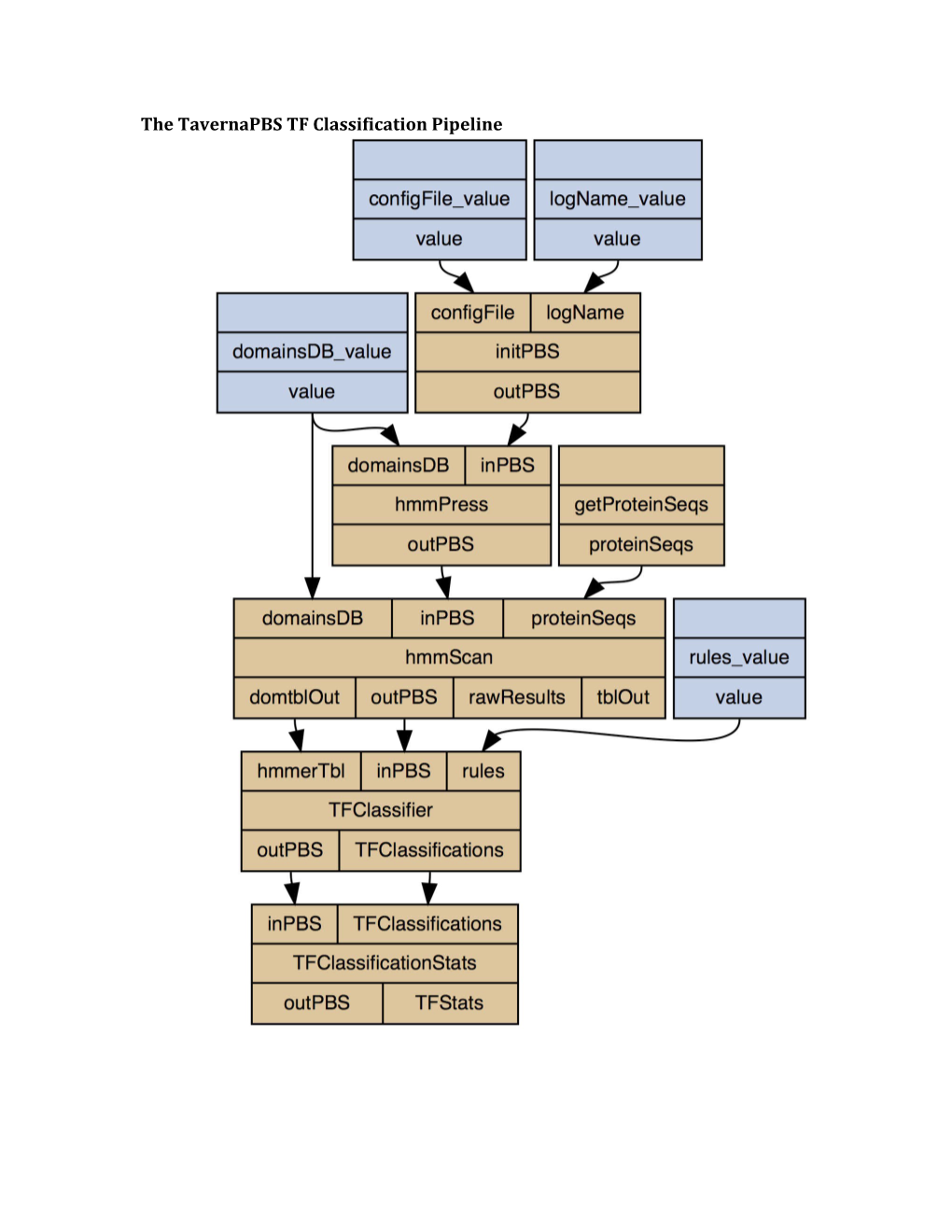
The TavernaPBS TF Classification Pipeline TF Classification SLURM Script
#!/bin/bash #SBATCH --partition=economy #SBATCH --account=timko-hiseq-analysis #SBATCH --nodes=1 #SBATCH --ntasks-per-node=4 #SBATCH --mem-per-cpu=4000 #SBATCH --time=48:00:00 cd $SLURM_SUBMIT_DIR for aa in *.fasta do /home/vam5g/hmmer-3.1b2-linux-intel-ia32/src/hmmpress -f /home/vam5g/TFClassification/domainsDB.hmm /home/vam5g/hmmer-3.1b2-linux-intel-ia32/src/hmmscan -o $aa.raw_results.txt --tblout $aa.tblout.txt --domtblout $aa.domtblout.txt --max --cpu 4 -E 1e-3 --domE 1e-3 /home/vam5g/TFClassification/domainsDB.hmm $aa /home/vam5g/TFClassification/TFRulesClassifier.pl /home/vam5g/TFClassification/TFRules.txt $aa.domtblout.txt > $aa.TFClassifications.txt /home/vam5g/TFClassification/TFRulesClassificationStats.pl $aa.TFClassifications.txt > $aa.TFClassifications.stats done
The code for tf-classifier_slurm.sh , a Bash script we designed for adapting the TavernaPBS pipeline for the SLURM cluster.
Eliminating Redundant Sequences
#!/usr/bin/perl use 5.010; use strict; use Bio::SeqIO; my %unique; my $file = $ARGV[0]; my $seqio = Bio::SeqIO->new(-file => $file, -format => "fasta"); my $outseq = Bio::SeqIO->new(-file => ">$file.unique.txt", -format => "fasta"); while(my $seqs = $seqio->next_seq) { my $id = $seqs->display_id; my $seq = $seqs->seq; unless(exists($unique{$seq})) { $outseq->write_seq($seqs); $unique{$seq} +=1; } }
The code for unique-sequences.pl , a BioPerl script we designed for ensuring that only unique amino acid sequences are included in the data set used for analyses.
DNA Translation in Six Frames
#!/usr/local/bin/perl use strict; use warnings; use lib '/Users/vikrammisra/.cpan/build/BioPerl-1.007001-1'; use Bio::SeqIO; use Bio::Seq; use Bio::DB::Fasta; use Bio::PrimarySeqI; my $input_file = $ARGV[0]; my $seqdb = Bio::DB::Fasta->new( $input_file ); my @ids = $seqdb->get_all_primary_ids; my $dna = ''; for ( my $ind = 0; $ind < scalar @ids; $ind++ ) { my $seqname = $ids[$ind]; chomp $seqname; $dna = $seqdb->seq($seqname); if ($dna =~ /[NnXx]/ ) { $dna =~ s/[NnXx]/\?/; } my $revseq = Bio::Seq->new(-seq=>$dna)->revcom;
my $prot = Bio::Seq->new(-seq=>$dna)->translate->seq; print `echo \Q>$seqname\_ORF_0F\E >> $input_file.aa.fasta `; print `echo $prot >> $input_file.aa.fasta`;
$prot = Bio::Seq->new(-seq=>$dna)->translate(-frame => 1)->seq; # $outprot = ">" . $seqname . "\n" . $prot . "\n"; # print `echo ">$seqname" >> proteins_1f.fasta`; print `echo \Q>$seqname\_ORF_1F\E >> $input_file.aa.fasta `; print `echo $prot >> $input_file.aa.fasta`; $prot = Bio::Seq->new(-seq=>$dna)->translate(-frame => 2)->seq; # $outprot = ">" . $seqname . "\n" . $prot . "\n"; print `echo \Q>$seqname\_ORF_2F\E >> $input_file.aa.fasta `; print `echo $prot >> $input_file.aa.fasta`;
$prot = $revseq->translate->seq; # $outprot = ">" . $seqname . "\n" . $prot . "\n"; print `echo \Q>$seqname\_ORF_0R\E >> $input_file.aa.fasta `; print `echo $prot >> $input_file.aa.fasta`;
$prot = $revseq->translate(-frame => 1)->seq; # $outprot = ">" . $seqname . "\n" . $prot . "\n"; print `echo \Q>$seqname\_ORF_1R\E >> $input_file.aa.fasta `; print `echo $prot >> $input_file.aa.fasta`;
$prot = $revseq->translate(-frame => 2)->seq; # $outprot = ">" . $seqname . "\n" . $prot . "\n"; print `echo \Q>$seqname\_ORF_2R\E >> $input_file.aa.fasta `; print `echo $prot >> $input_file.aa.fasta`;
}
The code for dna-translator_6frame.pl , a BioPerl script we designed to translate nucleotide sequences in six frames.
Separation into TF and TAP Families
#!/usr/bin/perl use 5.018;
# Remember to substitute the forward slash for underscore character in the PlantTFcat or other file # before you begin. use strict; use warnings; open NAMES, $ARGV[0] or die "Error in ARGV 0: $!"; #List of TF Family Names from a database such as PlantTFcat open TOSEARCH, $ARGV[1] or die "Error in ARGV 1: $!"; # Data file with TF classifications from that database for sequences studied print "What organism? "; my $organism =
The code for family_lines-from-list.pl , a Perl script we designed to separate TF sequence names into text files, each file having the name of the TF family in which the sequence is a member.
Sorting into TF Families, Accounting for Different Names (Synonyms) that Databases Use for the Same Family
#!/usr/bin/perl use strict; use warnings; use 5.012; open DBASE, $ARGV[0] or die "Error in ARGV 0: $!"; my $word; my $row; my $col; my @synonyms = (); while (
The code for tf-sorter.pl , a perl script we designed to sort TFs into their respective families, taking into account the synonyms that multiple databases have for the same family. For example, if one file of TFs has the name CCAAT_HAP2, and another file of TFs has the name NF-YA, the sequences from these files are sorted into a file with the name CCAAT_HAP2. Please note that this script is to be used after every TF sequence name is first sorted into its corresponding gene family for each pipeline and then stored in a text file bearing the name of that gene family. For example, all sequences belonging to the family “B3-Domain” from PlantTFcat should be stored in a file containing the name “B3-Domain”.
Eliminate Redundant Lines
#!/usr/bin/perl use strict; use warnings; open TOOBIG, $ARGV[0] or die "Can't open: $!"; open CLEANED, ">" . $ARGV[0] . "_unique.txt" or die "Can't write: $!"; my $stored; my @unique; while (
Code for the unique-lines.pl script, a Perl script we designed to eliminate redundant lines from a text file. The script tf-sorter.pl uses this script and needs to be in the same folder as unique-lines.pl .
TF Families and Synonyms
A20-like Alfin-like AP2_EREBP AP2_ERF-AP2AP2_ERF-ERFAP2_ERF-RAV RAV AP2-EREBP ARF B3-ARF Argonaute PAZ-Argonaute ARID ARID-HMG LOB AS2_LOB AS2-LOB AUX_IAA AUX-IAA B3 B3-Domain ABI3_VP1 BBR_BPC BBR-BPC GAGA-Binding-like BED-type_Zn BES_BZR BES1 bHLH bHSH Bromodomain BSD BTB-POZ bZIP bZIP1 bZIP2 C2C2_CO-like C2C2-CO-like C2C2_Dof C2C2-Dof C2C2_GATA C2C2-GATA C2C2_YABBY C2C2-YABBY C2C2-LSD Znf-LSD C2H2 C3H CAMTA CG1-CAMTA CCAAT_Dr1 CCAAT_HAP2 Hap2_NF-YA NF-YA CCAAT_HAP3 Hap3_NF-YB NF-YB CCAAT_HAP5 NF-YC CCHC_Zn CHROMO-DOMAIN Coactivator_p15 TCoAp15 ssDNA-binding-TF CPP Tesmin CSD CW-Zn CW-Zn-B3_VAL DBB Znf-B DBP DDT Dicer DUF246 DUF296 DUF547 DUF632 DUF833 E2F_DP E2F-DP EIL FAR FAR1 FHA FHA-SMAD FYR GARP_ARR-B_G2 GARP-ARR-B GARP_ARR-B_Myb RR-B-type GARP_G2-like GARP-G2-like GeBP GIF GNAT GRAS GRF C3H-WRC_GRF HB HB-KNOX HB_KNOX Homeodomain-TALE-KNOX HB-BELL Homeodomain-TALE-BEL HB-HD-ZIP HB-other Homeodomain-LIKE HB-PHD Homeodomain-PHD HB-WOX Homobox-WOX HD-SAD HD-ZIP HD-Zip_bZIP_1 HD-Zip_Halz HMG HRT HSA HSF HSF-type-DNA-binding ISWI IWS1 JmjC JmjC-ARID JmjN Jumonji KRAB-box Lambda-DB LFY LIM LUG LisH MADS MADS-M-typeMADS-MIKC MADS-type1 MBF1 MED6 MED7 mTERF MYB MYB_SANT MYB-HB-like MYB-related NAC NAM NF-X1 Nin-like RWP-RK None Others NOZZLE NZZ OFP PcG_FIE WD40-like PcG_VEFS PHD PLATZ Pseudo_ARR-B RR-A-type RB Rcd1-like Rel RF-X RRN3 Runt S1Fa-like SAP SBP SET Sigma70-like Sin3 Sir2 SNF2 SWI_SNF_SNF2 SOH1 SRS STY-LRP1 SSXT STAT SWI_SNF_BAF60b SWI_SNF-BAF60b SWI_SNF_SWI3 SWI_SNF-SWI3 SWIB-Plus-3 TAZ Tc-PD TCP TEA TFb2 Tify TRAF BTB-POZ-MATH ABTB Trihelix TTF-type_Zn TUB TUBBY ULT VARL VOZ Whirly WRKY YEATS zf_HD zf-HD
The list of synonyms for each family, used as a text file. Synonyms for the same family are separated by a tab.
TF Classification Pipeline Prioritization
#!/bin/sh
FILE=$1 # Sequence names with suffixes such as ORF+2 BASE=$2 # Sequence names without suffixes while read line ; do if ( ( ( grep -q "^$line$" $FILE ) || ( grep -q "$line.p" $FILE ) ) && ( ( grep -q "$line\_ORF[+-][123]" $FILE ) || ( grep -q "$line\-[012][FR]" $FILE ) || ( grep -q "$line\-[012][FR]" $FILE ) ) ) ; then grep -e "^$line$" -e "$line.p" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "^$line$" $FILE ) || ( grep -q "$line.p" $FILE ) ) ; then #Protein grep -e "^$line$" -e "$line.p" $FILE >> $FILE\_sorted.txt elif ( grep -q "$line\_ORF[+-][123]" $FILE ) ; then # PlantTFcat grep -e "$line\_ORF[+-][123]" $FILE >> $FILE\_sorted.txt elif ( grep -q "$line\-[012][FR]" $FILE ) ; then if ( ( grep -q "$line\-0F" $FILE ) && ! ( grep -q "$line\_ORF\+1" $FILE ) ) ; then grep -e "$line\-0F" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\-1F" $FILE ) && ! ( grep -q "$line\_ORF\+2" $FILE ) ) ; then grep -e "$line\-1F" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\-2F" $FILE ) && ! ( grep -q "$line\_ORF\+3" $FILE ) ) ; then grep -e "$line\-2F" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\-0R" $FILE ) && ! ( grep -q "$line\_ORF\-1" $FILE ) ) ; then grep -e "$line\-0R" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\-1R" $FILE ) && ! ( grep -q "$line\_ORF\-2" $FILE ) ) ; then grep -e "$line\-1R" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\-2R" $FILE ) && ! ( grep -q "$line\_ORF\-3" $FILE ) ) ; then grep -e "$line\-2R" $FILE >> $FILE\_sorted.txt fi elif ( grep -q "$line\_ORF\_[012][FR]" $FILE ) ; then if ( ( grep -q "$line\_ORF\_0F" $FILE ) && ! ( grep -q "$line\-0F" $FILE ) && ! ( grep -q "$line\_ORF\+1" $FILE ) ) ; then grep -e "$line\_\ORF\_0F" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\_ORF\_1F" $FILE ) && ! ( grep -q "$line\-1F" $FILE ) && ! ( grep -q "$line\_ORF\+2" $FILE ) ) ; then grep -e "$line\_\ORF\_1F" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\_ORF\_2F" $FILE ) && ! ( grep -q "$line\-2F" $FILE ) && ! ( grep -q "$line\_ORF\+3" $FILE ) ) ; then grep -e "$line\_\ORF\_2F" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\_ORF\_0R" $FILE ) && ! ( grep -q "$line\-0F" $FILE ) && ! ( grep -q "$line\_ORF\+1" $FILE ) ) ; then grep -e "$line\_\ORF\_0R" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\_ORF\_1R" $FILE ) && ! ( grep -q "$line\-1F" $FILE ) && ! ( grep -q "$line\_ORF\+2" $FILE ) ) ; then grep -e "$line\_\ORF\_1R" $FILE >> $FILE\_sorted.txt elif ( ( grep -q "$line\_ORF\_2R" $FILE ) && ! ( grep -q "$line\-2F" $FILE ) && ! ( grep -q "$line\_ORF\+3" $FILE ) ) ; then grep -e "$line\_\ORF\_2R" $FILE >> $FILE\_sorted.txt fi fi done < $BASE The script for tfdb-heirarchy.sh , a Bash script designed to prioritize TF classification schemes as specified in the Materials and Methods section.
MEME Results to FASTA Sequence Part 1 – MEME to Data Table
#!/usr/local/bin/perl use strict; use warnings; use 5.010; use lib '/Users/vikrammisra/.cpan/build/BioPerl-1.007001'; use Bio::SeqFeatureI; use Bio::AlignIO; use Bio::SimpleAlign; use Bio::Seq; use Bio::LocatableSeq; use Bio::SeqFeatureI; use Bio::FeatureHolderI; use List::MoreUtils qw(uniq); use Data::Dumper qw(Dumper); my $in_name = $ARGV[0]; open INFILE, '<', $in_name or die "Could not read $ARGV[0]: $!\n"; my $in = Bio::AlignIO->new(-file => $in_name, -format => 'meme'); open OUTFILE, '>' . $ARGV[0] . "-table.txt" or die "Could not write: $!\n";
# say OUTFILE "ID\tMotif Start\tMotif End\tAA Sequence"; while ( my $aln = $in->next_aln ) { foreach my $seqobj ($aln->each_seq()) { my $table_line = $seqobj->id . "\t" . $seqobj->start() . "\t" . $seqobj->end() . "\t" . $seqobj->seq; say OUTFILE $table_line; } } close INFILE; close OUTFILE;
The script for meme-to-table_aa.pl , a BioPerl script designed to take MEME motif results for amino acid sequences (and nucleotide sequences translated into six frames) and make a table of sequence names, motif start and end coordinates and found motif within the query sequence.
MEME Results to FASTA Sequence Part 2 – Data Table to FASTA
#!/usr/local/bin/perl use strict; use warnings; use 5.010; use List::MoreUtils qw(uniq); open INTABLE, '<' . $ARGV[0] or die "Could not read: $!"; open NEWFASTA, '>' . $ARGV[0] . "-results.fasta" or die "Could not write: $!"; my %starts; my %startset = (); my %seqset = (); my @nameset = (); while (
The script for memetable_to-fasta.pl , a Perl script designed to join MEME motifs in the order that they appear in the original query sequence, thus producing a curated amino acid sequence.
Remove Sequences with Greater than 95% Similarity
#!/usr/bin/perl use 5.010; use strict; use warnings; use Bio::SeqIO; use Bio::AlignIO; unless ( defined $ARGV[0] ) { die "Cannot open $ARGV[0]: $!"; } my $str = Bio::AlignIO->new(-file => $ARGV[0], -format => 'fasta'); my $out = Bio::AlignIO->new(-file => ">$ARGV[0].lt95.txt", -format => 'fasta'); my ($aln) = $str->next_aln(); $aln->purge(0.95); $out->write_aln($aln);
The code for purge95.pl , a BioPerl script we designed to remove sequences that are greater than 95% similar to each other, to ensure an accurate alignment.
BLAST Parser
#!/usr/bin/perl use 5.010; use strict; use Bio::SearchIO; my @conserved;
my $searchio = Bio::SearchIO->new( -format => 'blast', -file => $ARGV[0] ); while ( my $result = $searchio->next_result() ) { while( my $hit = $result->next_hit ) { while( my $hsp = $hit->next_hsp ) { print $result->query_name, "\t", $hit->name, "\n"; } } }
The code for blastparser.pl , a BioPerl script we designed to parse the BLASTN results from VuGEA, retrieving the query and the hit sequence name. Because the hit sequences were already listed from strongest homology to weakest homology, Microsoft Excel was used to take the two-column list of query and hit sequence, andremove redundant values in the query name column, leaving the query sequence name and the corresponding best hit sequence name.
Retrieve FPKM for a Transcript in VuGEA
#!/usr/bin/perl use 5.010; use strict; use warnings; open DATALIST, "<" . $ARGV[0] or die "Error in sequence file: $!"; open NAMELIST, "<" . $ARGV[1] or die "Error in name list: $!"; my %data_entries = (); while (
#foreach my $tester ( values %data_entries ) { #}
#foreach my $gridname ( @datanames ) { foreach my $category ( @subsets ) { foreach my $gridname ( @datanames ) { my $outfile = $category . "\\" . "_data.txt"; my @matches = grep /$gridname/, @{ $groups{$category} }; foreach my $val ( @matches ) { if ( $data_entries{$gridname} =~ /\d/ ) { my $output_line = $val . "\t" . $data_entries{$gridname}; my $finalform = `echo "$output_line" >> $outfile`; } else { next; } } } }
Code for fpkm-getter_vugea.pl , a Perl script we designed to retrieve the FPKM data for transcripts from VuGEA.
Preparing Heatmaps for a TF Family
#!/usr/bin/perl use 5.010; use strict; use warnings; open DATALIST, "<" . $ARGV[0] or die "Error in sequence file: $!"; open NAMELIST, "<" . $ARGV[1] or die "Error in name list: $!"; my %data_entries = (); while (
#foreach my $tester ( values %data_entries ) { #}
#foreach my $gridname ( @datanames ) { foreach my $category ( @subsets ) { foreach my $gridname ( @datanames ) { my $outfile = $category . "\\" . "_data.txt"; my @matches = grep /$gridname/, @{ $groups{$category} }; foreach my $val ( @matches ) { if ( $data_entries{$gridname} =~ /\d/ ) { my $output_line = $val . "\t" . $data_entries{$gridname}; my $finalform = `echo "$output_line" >> $outfile`; } else { next; } } } }
Code for search-data-by-group.pl , a Perl script we designed to take a two-column list with the first column being the cowpea genome sequence name and second column being the names of corresponding best-hit transcripts from VuGEA, and then use the VuGEA transcript name to find the FPKM data, and then create a table with the cowpea genome name in place of the VuGEA transcript name. This was done to prepare the FPKM data of a TF family for displaying on a tree in iTOL.
Making a Table with Gene, Genome Sequence and Data and Annotations for Best-Hit Transcript on VuGEA
#!/usr/bin/perl use 5.010; use strict; use warnings; open DATALIST, "<" . $ARGV[0] or die "Error in sequence file: $!"; open NAMELIST, "<" . $ARGV[1] or die "Error in name list: $!";
# Note: In NAMESET, the first column is assumed to be groups whereas the name # itself is in the second column. my %data_entries = (); while (
#foreach my $tester ( values %data_entries ) { # say $tester; #}
#foreach my $gridname ( @datanames ) { # foreach my $category ( @subsets ) { foreach my $category ( @subsets ) { foreach my $gridname ( @datanames ) { # my $outfile = $category . "\\" . "_data.txt"; my @matches = grep /$gridname/, @{ $groups{$category} }; foreach my $val ( @matches ) { # foreach my $val ( @{ $groups{$category} } ) { # if (( $val =~ /$gridname/ ) && ( $data_entries{$gridname} =~ /\d/ )) { # if ( ( $val =~ /$gridname/ ) && ( defined $data_entries{$gridname} ) ) { if ( $data_entries{$gridname} =~ /\d/ ) { say $category . "\t" . $val . "\t" . $data_entries{$gridname}; # say $output_line; # my $finalform = `echo "$output_line" >> $outfile`; } else { next; } } } } Code for annotation-table-prep.pl , a Perl script we designed to take the list of gene families and each member of these families, and then for each member, search against a table of VuGEA data, and then produce a file containing, for each genome sequence name, the best-hit VuGEA transcript, and the GO annotations and FPKM data for that transcript. Here the genome sequence name is the nucleotide sequence used to search against VuGEA, and its name does not contain any suffixes. This same script can then be used to treat the cowpea genome sequence and its data as two columns, and then retrieve the family for that genome sequence from a two-column list with the family as the first column and the genome sequences (which in this list has the suffixes such as “_ORF”).