Disruption of a Long-Range Cis-Acting Regulator for Shh Causes Preaxial Polydactyly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Novel ZRS Variant Causes Preaxial Polydactyly Type I by Increased Sonic Hedgehog Expression in the Developing Limb Bud
ARTICLE A novel ZRS variant causes preaxial polydactyly type I by increased sonic hedgehog expression in the developing limb bud Caixia Xu, PhD1, Xiaoming Yang, MD2,3, Hang Zhou, MD2,3, Yongyong Li, BS1, Chao Xing, PhD 4, Taifeng Zhou, MD2,3, Dongmei Zhong, BS1, Chengjie Lian, PhD2,3, Mei Yan, BS5, Tao Chen, BS5, Zhiheng Liao, MD2,3, Bo Gao, PhD6, Deying Su, BS2,3, Tingting Wang, MS2,3, Swarkar Sharma, PhD 7, Chandra Mohan, PhD8, Nadav Ahituv, PhD9,10, Sajid Malik, PhD 11, Quan-Zhen Li, PhD5 and Peiqiang Su, MD, PhD 2,3 Purpose: Preaxial polydactyly (PPD) is a common congenital hand Shh. We confirmed that HnRNP K can bind the ZRS and SHH malformation classified into four subtypes (PPD I–IV). Variants in promoters. The ZRS mutant enhanced the binding affinity for the zone of polarizing activity regulatory sequence (ZRS) within HnRNP K and upregulated SHH expression. LMBR1 intron 5 of the gene are linked to most PPD types. Conclusion: Our results identify the first PPD I disease-causing However, the genes responsible for PPD I and the underlying variant. The variant leading to PPD I may be associated with mechanisms are unknown. enhancing SHH expression mediated by HnRNP K. This study adds Methods: A rare large four-generation family with isolated PPD I to the ZRS-associated syndromes classification system for PPD and was subjected to genome-wide genotyping and sequence analysis. In clarifies the underlying molecular mechanisms. vitro and in vivo functional studies were performed in Caco-2 cells, 293T cells, and a knockin transgenic mouse model. -

Fly LMBR1/LIMR-Type Protein Lilipod Promotes Germ-Line Stem Cell Self-Renewal by Enhancing BMP Signaling
Fly LMBR1/LIMR-type protein Lilipod promotes germ-line stem cell self-renewal by enhancing BMP signaling Darin Dolezala,1, Zhiyan Liub,1, Qingxiang Zhoub, and Francesca Pignonia,b,c,2 aDepartment of Biochemistry and Molecular Biology, Upstate Medical University, Syracuse, NY 13210; bDepartment of Ophthalmology and Center for Vision Research, Upstate Medical University, Syracuse, NY 13210; and cDepartment of Neuroscience and Physiology, Upstate Medical University, Syracuse, NY 13210 Edited by Terry L. Orr-Weaver, Whitehead Institute, Cambridge, MA, and approved October 6, 2015 (received for review May 19, 2015) Limb development membrane protein-1 (LMBR1)/lipocalin-interact- cell remains in contact with the CCs and maintains stem cell ing membrane receptor (LIMR)-type proteins are putative nine- identity, whereas the other forms away from the niche and turns transmembrane receptors that are evolutionarily conserved across into a differentiating cystoblast (CB), the progenitor of egg cham- metazoans. However, their biological function is unknown. Here, we bers and ultimately oocytes. show that the fly family member Lilipod (Lili) is required for germ- The maintenance of ovarian stem cells is tightly regulated by line stem cell (GSC) self-renewal in the Drosophila ovary where it multiple extrinsic and intrinsic factors. Some of these factors re- enhances bone morphogenetic protein (BMP) signaling. lili mutant press the differentiation program in the renewed GSC, whereas GSCs are lost through differentiation, and display reduced levels of others relieve this repression in the CB. The major signaling sys- the Dpp transducer pMad and precocious activation of the master tem in this process is the BMP pathway (6). -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptJAMA Psychiatry Author Manuscript. Author Author Manuscript manuscript; available in PMC 2015 August 03. Published in final edited form as: JAMA Psychiatry. 2014 June ; 71(6): 657–664. doi:10.1001/jamapsychiatry.2014.176. Identification of Pathways for Bipolar Disorder A Meta-analysis John I. Nurnberger Jr, MD, PhD, Daniel L. Koller, PhD, Jeesun Jung, PhD, Howard J. Edenberg, PhD, Tatiana Foroud, PhD, Ilaria Guella, PhD, Marquis P. Vawter, PhD, and John R. Kelsoe, MD for the Psychiatric Genomics Consortium Bipolar Group Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis (Nurnberger, Koller, Edenberg, Foroud); Institute of Psychiatric Research, Department of Psychiatry, Indiana University School of Medicine, Indianapolis (Nurnberger, Foroud); Laboratory of Neurogenetics, National Institute on Alcohol Abuse and Alcoholism Intramural Research Program, Bethesda, Maryland (Jung); Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis (Edenberg); Functional Genomics Laboratory, Department of Psychiatry and Human Behavior, School of Medicine, University of California, Irvine (Guella, Vawter); Department of Psychiatry, School of Medicine, Corresponding Author: John I. Nurnberger Jr, MD, PhD, Institute of Psychiatric Research, Department of Psychiatry, Indiana University School of Medicine, 791 Union Dr, Indianapolis, IN 46202 ([email protected]). Author Contributions: Drs Koller and Vawter had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Nurnberger, Koller, Edenberg, Vawter. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: Nurnberger, Koller, Jung, Vawter. -

Selection Signatures Scan in Several Italian Sheep Breeds Identifies Genes Influencing Micronutrient Metabolism S
Selection signatures scan in several Italian sheep breeds identifies genes influencing micronutrient metabolism S. Sorbolini1, C. Dimauro1, M. Cellesi1, F. Pilla2, N.P.P. Macciotta1 and the BIOVITA Consortium. 1Università di Sassari, Dipartimento di Agraria, Viale Italia 39, 07100 Sassari, Italy. 1Università del Studi del Molise, Dipartimento Agricoltura Ambiente Alimenti, Via F. de Sanctis s.n.c. 86100 Campobasso, Italy. [email protected] (Corresponding author) Summary Animal physiological functions involve enzymes and cofactors. Many of these substances can not be synthesized by the body, but derive from the diet. An example are micronutrients, that strongly affect production performance, whose requirements are often met by the farmers by adding supplements to the diet. The aim of this study was to investigate the genetic variability of Italian sheep breeds searching for possible selective sweeps that harbor genes involved in the metabolism of micronutrients in. SNP A sample of 496 sheep belonging to 20 breeds farmed in Italy were genotyped with the Illumina Ovine 50K beadchip. Data were analysed by canonical discriminant analysis (CDA). Forty SNP located in regions of the genome containing loci involved in the metabolism of vitamins and minerals were detected. in particular, genes linked to the metabolism of vitamins and minerals such as Selenocysteine Lyase (SCLY), calcium sensing receptor (CASR), Solute Carrier Family 23 Member 1 (SLC23A1) and Thiamine Triphosphatase (THTPA) were highlighted. Keywords: selection signatures, micronutrients, sheep, Canonical Discriminant Analysis Introduction Nutritional status is of particular importance for productive performances in livestock. Growth, milk production, reproduction depend on a wide range of essential nutrients such as amino acids, fatty acids, vitamins and minerals. -

A Novel ZRS Mutation Leads to Preaxial Polydactyly Type 2 in a Heterozygous Form and Werner Mesomelic Syndrome in a Homozygous Form Julia E
A Novel ZRS Mutation Leads to Preaxial Polydactyly Type 2 in a Heterozygous Form and Werner Mesomelic Syndrome in a Homozygous Form Julia E. VanderMeer, University of California Reymundo Lozano, UC Davis Medical Center Miao Sun, Johns Hopkins University Yuan Xue, Emory University Donna Daentl, Shriners’ Hospitals for Children Ethylin Wang Jabs, Johns Hopkins University William R. Wilcox, Emory University Nadav Ahituv, University of California Journal Title: Human Mutation Volume: Volume 35, Number 8 Publisher: Wiley: 12 months | 2014-08, Pages 945-948 Type of Work: Article | Post-print: After Peer Review Publisher DOI: 10.1002/humu.22581 Permanent URL: http://pid.emory.edu/ark:/25593/gjzd4 Final published version: http://onlinelibrary.wiley.com/doi/10.1002/humu.22581/abstract;jsessionid=A81B9AAB2E2A3B4055E6A9A0004D6BC3.f03t04?systemMessage=Wiley+Online+Library+will+be+disrupted+9th+Aug+from+10-2+BST+for+essential+maintenance.+Pay+Per+View+will+be+unavailable+from+10-6+BST. Copyright information: © 2014 Wiley Periodicals, Inc. Accessed September 26, 2021 9:00 PM EDT NIH Public Access Author Manuscript Hum Mutat. Author manuscript; available in PMC 2014 August 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Hum Mutat. 2014 August ; 35(8): 945–948. doi:10.1002/humu.22581. A Novel ZRS Mutation Leads to Preaxial Polydactyly Type 2 in a Heterozygous Form and Werner Mesomelic Syndrome in a Homozygous Form Julia E. VanderMeer1,2,†, Reymundo Lozano3,†, Miao Sun4,5,†, Yuan Xue6, Donna -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Physical Mapping of QTL for Tuber Yield
Schönhals et al. BMC Genomics (2017) 18:642 DOI 10.1186/s12864-017-3979-9 RESEARCH ARTICLE Open Access Physical mapping of QTL for tuber yield, starch content and starch yield in tetraploid potato (Solanum tuberosum L.) by means of genome wide genotyping by sequencing and the 8.3 K SolCAP SNP array Elske Maria Schönhals1, Jia Ding1, Enrique Ritter2, Maria João Paulo3, Nicolás Cara1, Ekhard Tacke4, Hans-Reinhard Hofferbert5, Jens Lübeck6, Josef Strahwald6 and Christiane Gebhardt1* Abstract Background: Tuber yield and starch content of the cultivated potato are complex traits of decisive importance for breeding improved varieties. Natural variation of tuber yield and starch content depends on the environment and on multiple, mostly unknown genetic factors. Dissection and molecular identification of the genes and their natural allelic variants controlling these complex traits will lead to the development of diagnostic DNA-based markers, by which precision and efficiency of selection can be increased (precision breeding). Results: Three case-control populations were assembled from tetraploid potato cultivars based on maximizing the differences between high and low tuber yield (TY), starch content (TSC) and starch yield (TSY, arithmetic product of TY and TSC). The case-control populations were genotyped by restriction-site associated DNA sequencing (RADseq) and the 8.3 k SolCAP SNP genotyping array. The allele frequencies of single nucleotide polymorphisms (SNPs) were compared between cases and controls. RADseq identified, depending on data filtering criteria, between 6664 and 450 genes with one or more differential SNPs for one, two or all three traits. Differential SNPs in 275 genes were detected using the SolCAP array. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -

Genome Provides Insights Into Vertebrate Evolution
ARTICLES OPEN Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution Jeramiah J Smith1,2, Shigehiro Kuraku3,4, Carson Holt5,37, Tatjana Sauka-Spengler6,37, Ning Jiang7, Michael S Campbell5, Mark D Yandell5, Tereza Manousaki4, Axel Meyer4, Ona E Bloom8,9, Jennifer R Morgan10, Joseph D Buxbaum11–14, Ravi Sachidanandam11, Carrie Sims15, Alexander S Garruss15, Malcolm Cook15, Robb Krumlauf15,16, Leanne M Wiedemann15,17, Stacia A Sower18, Wayne A Decatur18, Jeffrey A Hall18, Chris T Amemiya2,19, Nil R Saha2, Katherine M Buckley20,21, Jonathan P Rast20,21, Sabyasachi Das22,23, Masayuki Hirano22,23, Nathanael McCurley22,23, Peng Guo22,23, Nicolas Rohner24, Clifford J Tabin24, Paul Piccinelli25, Greg Elgar25, Magali Ruffier26, Bronwen L Aken26, Stephen M J Searle26, Matthieu Muffato27, Miguel Pignatelli27, Javier Herrero27, Matthew Jones6, C Titus Brown28,29, Yu-Wen Chung-Davidson30, Kaben G Nanlohy30, Scot V Libants30, Chu-Yin Yeh30, David W McCauley31, James A Langeland32, Zeev Pancer33, Bernd Fritzsch34, Pieter J de Jong35, Baoli Zhu35,37, Lucinda L Fulton36, Brenda Theising36, Paul Flicek27, Marianne E Bronner6, All rights reserved. Wesley C Warren36, Sandra W Clifton36,37, Richard K Wilson36 & Weiming Li30 Lampreys are representatives of an ancient vertebrate lineage that diverged from our own ~500 million years ago. By virtue of this deeply shared ancestry, the sea lamprey (P. marinus) genome is uniquely poised to provide insight into the ancestry of vertebrate genomes and the underlying principles of vertebrate biology. Here, we present the first lamprey whole-genome sequence and America, Inc. assembly. We note challenges faced owing to its high content of repetitive elements and GC bases, as well as the absence of broad-scale sequence information from closely related species. -

Whole Genome Resequencing Reveals an Association of ABCC4 Variants with Preaxial Polydactyly in Pigs Cheng Ma1,2,3, Saber Khederzadeh1,2,3, Adeniyi C
Ma et al. BMC Genomics (2020) 21:268 https://doi.org/10.1186/s12864-020-6690-1 RESEARCH ARTICLE Open Access Whole genome resequencing reveals an association of ABCC4 variants with preaxial polydactyly in pigs Cheng Ma1,2,3, Saber Khederzadeh1,2,3, Adeniyi C. Adeola1, Xu-Man Han1, Hai-Bing Xie1* and Ya-Ping Zhang1* Abstract Background: Polydactyly is one of the most common congenital limb dysplasia in many animal species. Although preaxial polydactyly (PPD) has been comprehensively studied in humans as a common abnormality, the genetic variations in other animal species have not been fully understood. Herein, we focused on the pig, as an even-toed ungulate mammal model with its unique advantages in medical and genetic researches, two PPD families consisting of four affected and 20 normal individuals were sequenced. Results: Our results showed that the PPD in the sampled pigs were not related to previously reported variants. A strong association was identified at ABCC4 and it encodes a transmembrane protein involved in ciliogenesis. We found that the affected and normal individuals were highly differentiated at ABCC4, and all the PPD individuals shared long haplotype stretches as compared with the unaffected individuals. A highly differentiated missense mutation (I85T) in ABCC4 was observed at a residue from a transmembrane domain highly conserved among a variety of organisms. Conclusions: This study reports ABCC4 as a new candidate gene and identifies a missense mutation for PPD in pigs. Our results illustrate a putative role of ciliogenesis process in PPD, coinciding with an earlier observation of ciliogenesis abnormality resulting in pseudo-thumb development in pandas. -
Primepcr™Assay Validation Report
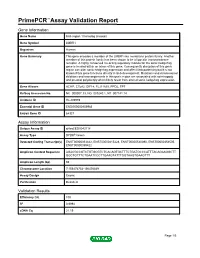
PrimePCR™Assay Validation Report Gene Information Gene Name limb region 1 homolog (mouse) Gene Symbol LMBR1 Organism Human Gene Summary This gene encodes a member of the LMBR1-like membrane protein family. Another member of this protein family has been shown to be a lipocalin transmembrane receptor. A highly conserved cis-acting regulatory module for the sonic hedgehog gene is located within an intron of this gene. Consequently disruption of this genic region can alter sonic hedgehog expression and affect limb patterning but it is not known if this gene functions directly in limb development. Mutations and chromosomal deletions and rearrangements in this genic region are associated with acheiropody and preaxial polydactyly which likely result from altered sonic hedgehog expression. Gene Aliases ACHP, C7orf2, DIF14, FLJ11665, PPD2, TPT RefSeq Accession No. NC_000007.13, NG_009240.1, NT_007741.14 UniGene ID Hs.209989 Ensembl Gene ID ENSG00000105983 Entrez Gene ID 64327 Assay Information Unique Assay ID qHsaCED0043118 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000353442, ENST00000415428, ENST00000540390, ENST00000354505, ENST00000359422 Amplicon Context Sequence AGACGCCGTCTGTGCGTCTCACAGTGCTTTCTGATGCCCATTTACAGAAGGCTT GGCTGTTTCTGAATCCCTTGAAGTATTTGGTAAGTGAAGTTT Amplicon Length (bp) 66 Chromosome Location 7:156476754-156476849 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9994 cDNA Cq 21.19 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 77.5 gDNA Cq 24.55 Specificity (%) 100 -

Extensive Viral Mimicry of Human Proteins in AIDS, Multiple Sclerosis and Other Autoimmune Disorders, Late-Onset and Familial Al
Extensive Viral mimicry of human proteins in AIDS, multiple sclerosis and other autoimmune disorders, late-onset and familial Alzheimer’s disease and other genetic diseases C.J.Carter Flat 4, 20 Upper Maze Hill, St Leonard’s on Sea, East Sussex, TN38 OLG [email protected] Abstract Peptide stretches within HIV-1 proteins display a striking homology to over 50 important components of the human immune and pathogen defence network. These include HLA-antigens, T-cell, Fc and cytokine receptors, CD molecules, lymphocyte antigens, proteins involved in B-Cell, T-cell, dendritic and natural killer cell, macrophage, mast cell and microglial function, lysosomal proteins, haematopoietic control, and also pathogen recognition pathways. The homologous peptides are in most cases highly immunogenic (B-cell epitope prediction), suggesting that antibodies to HIV-1 proteins could mount an autoimmune attack against multiple components of the immune system itself. HIV-1 proteins are also homologous to autoantigens in Alzheimer’s disease, chronic obstructive pulmonary disorder, multiple sclerosis, Myasthenia Gravis, Pemphigus Vulgaris, Sjogrens syndrome and systemic Lupus Erythematosus, all of which have been associated with HIV-1 infection. This mimicry suggests that HIV-1/AIDS has a major autoimmune component and that HIV-1 antibodies could selectively target the immune system and autoantigens in other autoimmune disorders. This could radically change our conception of how HIV- 1 acts, and perhaps lead to novel therapeutic strategies, which, counter intuitively might even involve the use of immunosuppressants in the early stages of the disease. Autoantigens from the human autoimmune diseases mentioned above also align with peptides from other viruses implicated as risk factors in each disease.